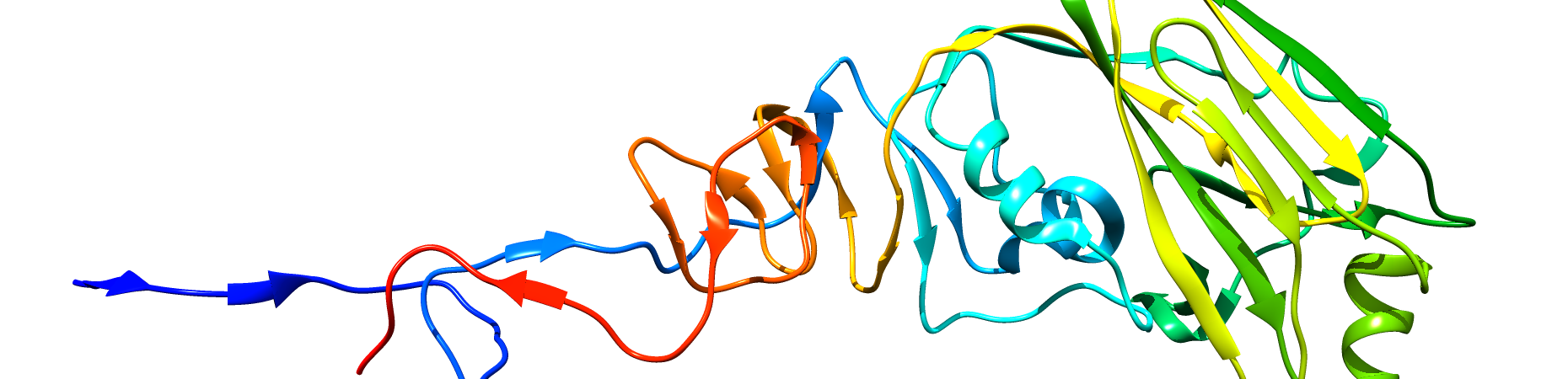
PROTEIN
Influenza virus Hemagglutinin, chain alpha.
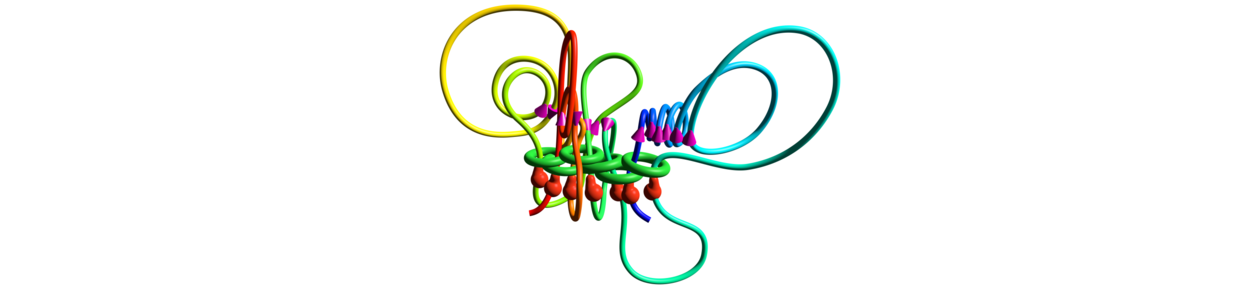
GENE
3D representation of a topological domain mediated by CTCF (red bulbs), cohesin (green rings) and the embedded transcriptional complex (purple cones). This model is an average representation of data obtained from millions of cells.

GENOME
Model of megabase sized region of human Chromosome 7 obtained from 3D-GNOME Monte Carlo webserver using ChIA-PET data.
CELL
Cancer cells colony. Picture from the simulation of growing breast cancer tumor.
Project TEAM - Laboratory of Functional and Structural Genomics CeNT
Three-dimensional Human Genome structure at the population scale: computational algorithm and experimental validation for lymphoblastoid cell lines of selected families from 1000 Genomes Project
RESEARCH
STRUCTURAL GENOMICS
Modeling of the 3D structure of chromatin inside the cell nucleus based on the ChIA-PET and Hi-C data.
FUNCTIONAL GENOMICS
Studying how different genomic regions interact and cooperate together in order to gain better understanding of the genotype-phenotype relationship.

BIOSTATISTICS AND COGNITIVE COMPUTING
Field of expertise: prediction, machine learning, statistics, computational methods, data preparation.
BIOLOGICAL SYSTEMS MODELING
Within the framework of iCell project we develop the novel computational modelling framework for multiscale analysis of tumor growth.
SERVICES

TEAM

Dariusz Plewczyński
lab leader
PhD DSc
Dariusz Plewczynski, PhD, is an assistant professor at University of Warsaw, the head of Functional and Structural Laboratory in Center of New Technologies CeNT, Warsaw, Poland. His expertise covers functional and structural genomics with the special focus on higher-order chromatin organization and its impact on gene regulation and transcription. The major computational tools that are used in his interdisciplinary research team include statistical data analysis (GWAS studies, clustering, machine learning), genomic variation analysis using diverse data sources (karyotyping, confocal microscopy, aCGH microarrays, next generation sequencing: both whole genome and whole exome), bioinformatics (protein sequence analysis, protein structure prediction), and finally biophysics (polymer theory and simulations) and genomics (epigenetics, genome domains, three dimensional structure of chromatin).
Tel: (+48 22) 55 43 654
E-mail: d.plewczynski(at)cent.uw.edu.pl

Michał Łaźniewski
researcher
PhD
Michał Łaźniewski scientific interest focus mostly on proteins their sequence, structure, function and interactions with small drug-like compounds. Constant progress in sequencing techniques results, among others, in the exponential growth of the number of known protein sequences. Using profile and meta-profile methods this new sequences can be linked to other alre ady characterized proteins. These information can be used to group similar proteins, predict the structure of newly identified ones and ,to some extent, predict their function. These tasks can be achieved even if compared object share only marginal sequence similarity. Using protein structure a number of analysis can be performed, protein dynamics, predicting protein-protein interactions or calculating binding affinities of small molecules to interesting proteins. These analysis can help us to better understand biological processes at the molecular level and to to predict new drugs.
E-mail: m.lazniewski(at)cent.uw.edu.pl
Research areas: FUNCTIONAL GENOMICS

Przemysław Szałaj
researcher
PhD candidate
Przemek Szałaj is interested in the functional and structural genomics, with a particular interest in the 3D chromatin organization. He developed simulation software for inferring the multiscale chromatin models using Hi-C and ChIA-PET data. Currently he works on applying the bioimaging data for refinement of the simulations. He is also interested in the variation of the structural genomic properties and their inheritance, with the ultimate goal being to better understand the significance of the genome folding on its function and the mechanisms that govern it.
E-mail: p.szalaj(at)cent.uw.edu.pl
Research areas: STRUCTURAL GENOMICS

Weronika Wronowska
researcher
MSc
Weronika Wronowska interests are focused on Systems Biology. In particular in the area of her interest is mathematical modeling of life processes and functional analysis of high through-put proteomic, transcriptomic and lipidomic data. She was involved in the project in collaboration with MD Anderson Cancer Center investigating heterogeneity of gene expression profile of bladder cancer subtypes. In which thanks to a comprehensive analysis of transcriptomic data, it managed to establish markers of tumor progression. Currently, her research focuses on a formal modeling of the sphingolipid metabolism. In particular the role of this bioactive lipids in progression of human disease like cancer and neurodegenerative disorders. In this field a variety of modeling approaches are used, such as deterministic modeling using different types of kinetics and constraint based large-scale modeling of whole cellular metabolism. She is also interested in developing of new tools for integration of high through-put data with formal models. Her two main goals are (i) development of a global cellular metabolism model of high predictive value, and (ii) dissemination of mathematical models as tools for medical diagnosis and the design of new drugs.
E-mail: w.wronowska(at)cent.uw.edu.pl
Research areas: BIOLOGICAL SYSTEMS MODELING

Michał Sadowski
researcher
BSc
Michał Sadowski interests are from the field of physics and genome biology and are presently focused on genome architecture, genome organization and their link to gene regulation. These areas of study can be explored by an analysis of data coming from experimental methods which capture chromatin conformation both locally (3C, 4C) and genome-wide (Hi-C and ChIA-PET). This analysis can lead us to better understanding of gene expression mechanisms and provide a new insight into relation between gene expression and genomic variation. In his present work Michał Sadowski is trying to combine conformation capture data, genomic variation data and gene transcription data, in order to pursue more complete explanation of gene regulation processes, genome spatial arrangement and its evolution.
E-mail: m.sadowski(at)cent.uw.edu.pl
Research areas: STRUCTURAL GENOMICS

Grzegorz Bokota
researcher
MSc
Grzegorz Bokota interests are focused on developing algorithm for microscope image analyzing and massive parallel modeling of cell colony. In image analysis created algorithm are focused on nuclei segmentation and nuclei analysis of the interior of the nucleus.
E-mail: g.bokota(at)cent.uw.edu.pl
Research areas: STRUCTURAL GENOMICS , BIOLOGICAL SYSTEMS MODELING

Teresa Szczepińska
researcher
PhD
Teresa Szczepińska, PhD, is a bioinformatician. She has bachelor’s degree in molecular biology from Interfaculty Individual Studies in Mathematical and Natural Sciences at Warsaw University and master’s degree in bioinformatics from VU University Amsterdam. She accomplished doctoral degree at Nencki Institute of Experimental Biology, Polish Academy of Sciences. Teresa’s research interest is in genomic data, higher order chromatin organisation and its relation to transcription regulation. She is experienced in high throughput sequencing data analysis and in retrieving of biological information with it’s knowledge-based selection.
E-mail: t.szczepinska(at)cent.uw.edu.pl
Research areas: STRUCTURAL GENOMICS

Michał Denkiewicz
researcher
PhD candidate
Reasercher with degrees in experimental cognitive psychology and computer science. Strong background in statistics and machine learning. Primary research area include mathematical modeling of problem solving and decision making in small groups. Scientific intrests: multiagent systems, social and embodied cognition, theory of cognitive systems.
E-mail: m.denkiewicz(at)cent.uw.edu.pl
Research areas: BIOSTATISTICS AND COGNITIVE COMPUTING

Wayne Dawson
researcher
PhD
Wayne Dawson, PhD, is a physicist and structural biologist. He has been a postdoc in the Laboratory of Functional and Structural Genomics since April 2016. He received his PhD from The University of Tokyo. His main focus over the years has been on RNA/protein structure prediction and folding, electron transfer proteins, and currently, chromatin structure and problems related to the influenza virus.
E-mail: dawsonzhu(at)gmail.com
Research areas: STRUCTURAL GENOMICS

Michal Pietal
researcher
Executive DBA, PhD
Michal was researching transformations of chromatin 2D interaction matrices (Hi-C / ChIA-PET) into 3D models or ensembles, with the use of various 2D processing techniques, including fuzzy graph distance maps and Multi-dimensional Scaling (MDS). He was also involved into Hemagglutinin molecule de novo modelling, with the use of fuzzy contact maps of homologous templates from PDB. In addition, he was engaged in protein-protein interface classification from sequence data, with 2D maps prediction stage.
Research areas: STRUCTURAL GENOMICS

Anna Maria Rusek
intern
PhD student
Anna Maria Rusek graduated Silesian University as a Master of Science in Biotechnology and completed postgraduate studies at Jagielonian University in the area of Laboratory Medicine. Currently she is a PhD student at Interdisciplinary Doctoral Studies in English at Medical University of Bialystok in cooperation with CeNT and in the process of caring out specialization in the field of Laboratory Medical Genetics in Łódź. The main field of interests is medical biology, in particular cancer signalling pathways and microRNAs involvement in cancer progression and molecular medicine, especially mechanisms of novel gene and immune therapies as well as molecular aspects of resistance to existing therapies.
Research areas: BIOLOGICAL SYSTEMS MODELING

Denis Kazakievich
researcher
PhD student
Denis is a PhD student at joint interdisciplinary PhD program at Uniwersytet Medyczny w Białymstoku and Universiteit Hasselt. He is working to construct statistical models for RNA-Seq data analysis and to develop practical applications of whole-cell modeling . He earned MEng in Bioinformatics from Politechnika Wrocławska and MD from Medical University of Grodno.
Research areas: BIOLOGICAL SYSTEMS MODELING

Ziad Al Bkhetan
researcher
PhD student
Ziad Al Bkhetan is a PhD student at University of Warsaw, He obtained his master degree in computer science from Warsaw University of Technology, Faculty of Mathematics and Information Science. He has good experience in Software Engineering, Data Mining and GPU Programming. Currently he is working on Genomic Interactions Prediction.
E-mail: z.albkhetan(at)cent.uw.edu.pl
Research areas: STRUCTURAL GENOMICS , BIOSTATISTICS AND COGNITIVE COMPUTING

Michał Kadlof
researcher
PhD student
Michał Kadlof graduated with a degree in bioinformatics and engineering studies in computer science with a specialization in database engineering. Currently he is doing a PhD in the Faculty of Physics at the University of Warsaw. His main interests are in computer simulations of the dynamics of biopolymers - proteins and nucleic acids. In the past, he dealt with protein structure prediction, and topology of proteins. Currently is conducting research on multiscale force fields that describe the behavior of chromatin within the nucleus. In addition, he is a computer systems administrator. He spends his free time doing electronics, hiking and medieval historical reenactments.
Tel: (+48 22) 55 43 752
E-mail: m.kadlof(at)cent.uw.edu.pl
Research areas: STRUCTURAL GENOMICS

Paulina Urban
researcher
PhD student
Paulina Urban is a PhD student at The Inter-Faculty Studies in Mathematics and Natural Science at Warsaw University. Her supervisors are professor Jan Karbowski and professor Dariusz Plewczyński. She is a biologist and neuroinformatics. The main field of interests is signal and image analysis.
Research areas: BIOSTATISTICS AND COGNITIVE COMPUTING , BIOLOGICAL SYSTEMS MODELING

Zofia Parteka
researcher
MSc student
Zofia Parteka interests focus mostly on 3D structure modelling and combining structural and functional genomic data. She is currently working on her masters degree in the field of Bioinformatics and Systems Biology. In her current work she is trying to use ChIA-PET data in 3D chromatin modelling.

Agnieszka Kraft
researcher
MSc student
Agnieszka Kraft is a student at the Bioinformatics and Systems Biology master's programme. Her interests are in genome architecture, especially structural variants and their link to phenotypes.
Research areas: STRUCTURAL GENOMICS

Karolina Jodkowska
researcher
PhD
Karolina Jodkowska completed her BSc and MSc in Biology at the University of Warsaw. She did her PhD at the Spanish National Cancer Research Centre (CNIO) in Madrid studying spatial organization of DNA replication in the nucleus. Her interests are focused on understanding how basic cellular processes such as replication, transcription and DNA repair are coordinated in the context of three-dimensional organization of chromatin

Natalia Zawrotna
researcher
MSc student
Natalia Zawrotna is a student of Molecular Biology master's program at the University of Warsaw. She is interested in the functional and structural genomics. The main field of interests is long-range chromatin interactions and spatial genome organisation. She is a member of the TEAM. In collaboration with other members they application to experimental work in NGS-based 3D genomics techniques: Hi-C, ChIA-PET and HiChIP (chromatin conformation capture).
Tel: (+48 22) 55 43 678
E-mail: n.zawrotna@cent.uw.edu.pl

Anup Kumar Halder
researcher
PhD student
Anup Kumar Halder is a Visvesvaraya PhD Fellow in the department of Computer Science and Engineering, Jadavpur University, India under the guidance of Dr. Subhadip Basu. He received his Bachelor of Technology degree in Computer Science and Engineering from Bankura Unnayani Institute of Engineering, Bankura, India in 2011 and Masters of Engineering degree in Computer Science and Engineering from Jadavpur University, Kolkata, India in 2014. He received Ministry of Human Resource Development (MHRD),GOI sponsored GATE scholarship for his Post Graduate degree. His area of current research interest is Computational Biology and Bioinformatics.He developed clustering algorithm for large biological sequences, predictive system for Protein Protein Interactions and interaction network analysis.

Andrzej Szczepańczyk
researcher
MSc student
Andrzej Szczepańczyk studied at Military University of Technology in cybernetics departement, IT faculty in 2012-2016. He is a C/C++ and Python programmer. He is currently working on his masters degree at ICM since October 2018r. In his current work he is working on 3D modelling microscopy data. He is also interested in mathematical modelling. He spends his free time singing and solving mathematical puzzles.

Anna Bugaj
researcher
MSc student
Student at Cognitive Science master’s programme, interested in molecular biology of cognitive and mental disorders, particularly Alzheimer’s Disease, Depression and Autism Spectrum Disorders. Working on master thesis exploring Structural Variants and SNPs from ASD in context of chromatin architecture.
Read our lab info
In the Laboratory of Functional and Structural Genomics we perform theoretical studies, whose main objective is to analyze and predict the three-dimensional structure of the human genome, and its relation with the genomic diversity of human populations, both natural and pathological. In particular, we investigate structural variants, copy number variants observed in various sub-populations and the groups of patients, and their three-dimensional localization in the structure of the nucleus.
We also examine the relationship of the expression levels of selected genes from their location in three-dimensional space. In addition, we use structural information to enrich the sequential genomic analysis in order to better define the function of selected genomic regions that are important in the context of personalized medicine.
For this purpose, first we are developing a variety of large-scale computational tools for analysis of whole genome sequences, the identification of structural variants, determining the statistical significance of the observed number of copies of genomic regions in selected cohorts of patients. Secondly, we evaluate their uniqueness comparing the observed changes with typical and natural genomic diversity that has been cataloged for example in the 1000 Genomes Project Consortium. Thirdly, we infer the biological function of these genomic regions using publicly available databases. Fourthly, we identify unique local three-dimensional environment for selected sites, eg. regulatory ones. In the fifth step, we analyze the impact of structural re-arrangements of those local neighborhoods on the gene expression profiles, which is related to the presence of transcription factories.
PAPERS
STRUCTURAL GENOMICS
2016
Abstract: Recent advances in high-throughput chromosome conformation capture (3C) technology, such as Hi-C and ChIA-PET, have demonstrated the importance of 3D genome organization in development, cell differentiation and transcriptional regulation. There is now a widespread need for computational tools to generate and analyze 3D structural models from 3C data. Here we introduce our 3D GeNOme Modeling Engine (3D-GNOME), a web service which generates 3D structures from 3C data and provides tools to visually inspect and annotate the resulting structures, in addition to a variety of statistical plots and heatmaps which characterize the selected genomic region. Users submit a bedpe (paired-end BED format) file containing the locations and strengths of long range contact points, and 3D-GNOME simulates the structure and provides a convenient user interface for further analysis. Alternatively, a user may generate structures using published ChIA-PET data for the GM12878 cell line by simply specifying a genomic region of interest. 3D-GNOME is freely available at http://3dgnome.cent.uw.edu.pl/.
Authors: SzalajP, Michalski PJ, Wróblewski P, Tang Z, Kadlof M, Mazzocco G, Ruan Y, Plewczynski D
Note: '3D-GNOME: an integrated web service for structural modeling of the 3D genome' by SzalajP, Michalski PJ, Wróblewski P, Tang Z, Kadlof M, Mazzocco G, Ruan Y, Plewczynski D. NucleicAcids Res. 2016 May 16. pii: gkw437. pmid:27185892
2015
Authors: Tang Z, Luo OJ, Li X, Zheng M, Zhu JJ, Szalaj P, Trzaskoma P, Magalska A, Wlodarczyk J, Ruszczycki B, Michalski P, Piecuch E, Wang P, Wang D, Tian SZ, Penrad-Mobayed M, Sachs LM, Ruan X, Wei CL, Liu ET, Wilczynski GM, Plewczynski D, Li G, Ruan Y
Note: 'CTCF-Mediated Human 3D Genome Architecture Reveals Chromatin Topology for Transcription' Tang Z, Luo OJ, Li X, Zheng M, Zhu JJ, Szalaj P, Trzaskoma P, Magalska A,Wlodarczyk J, Ruszczycki B, Michalski P, Piecuch E, Wang P, Wang D, Tian SZ, Penrad-Mobayed M, Sachs LM, Ruan X, Wei CL, Liu ET, Wilczynski GM, Plewczynski D, Li G, Ruan Y.Cell 2015, Dec 17;163(7):1611-27. doi: 10.1016/j.cell.2015.11.024. Epub 2015 Dec 10.
2013
Abstract: 3D-Hit is a well established method for rapid detection of structural similarities between proteins, which is widely used in various bioinformatics web servers (MetaServer, GRDB, 3D-Fun, Rosetta, etc.). The algorithm decomposes proteins into set of overlaping segments of 9–13 residues, then tries to match them using root mean square distance metric. The best aligned pairs of segments are selected as seeds for futher analysis. Those initial hits are expanded by iterative process in order to construct the global structural alignment by concatenating pairs of matching segments. The method has the same accuracy as the other state-of-the-art structural comparison algorithms (LGscore2, DALI), yet it provides much faster processing times, and can be used in a high-throughput setup as the structural module of bioinformatics pipelines. The method is optimized in terms of speed and accuracy to work on novel computer architectures, such as PowerXCell8i and Sun Constellation System. Here, we provide the source code of the 3D-Hit program, describe selected architectures on which the software was ported, present programing models, point out significant porting steps and sumarize performance comparisons.
Authors: Ł Bieniasz-Krzywiec, Maciej Cytowski, L Rychlewski and D Plewczynski
Note: '3D-Hit: fast structural comparison of proteins on multicore architectures' by Ł. Bieniasz-Krzywiec, Maciej Cytowski, L. Rychlewski and D. Plewczynski. Optimization Letters (2013).
FUNCTIONAL GENOMICS
2016
Abstract: We report the sequences of 1,244 human Y chromosomes randomly ascertained from 26 worldwide populations by the 1000 Genomes Project. We discovered more than 65,000 variants, including single-nucleotide variants, multiple-nucleotide variants, insertions and deletions, short tandem repeats, and copy number variants. Of these, copy number variants contribute the greatest predicted functional impact. We constructed a calibrated phylogenetic tree on the basis of binary single-nucleotide variants and projected the more complex variants onto it, estimating the number of mutations for each class. Our phylogeny shows bursts of extreme expansion in male numbers that have occurred independently among each of the five continental superpopulations examined, at times of known migrations and technological innovations.
Authors: Poznik GD, Xue Y, Mendez FL, Willems TF, Massaia A, Wilson Sayres MA, Ayub Q, McCarthy SA, Narechania A, Kashin S, Chen Y, Banerjee R, Rodriguez-Flores JL, Cerezo M, ShaoH, Gymrek M, Malhotra A, Louzada S, Desalle R, Ritchie GR, Cerveira E, Fitzgerald TW, Garrison E, Marcketta A, Mittelman D, Romanovitch M, Zhang C, Zheng-Bradley X, Abecasis GR, McCarroll SA, Flicek P, Underhill PA, Coin L, Zerbino DR, Yang F, Lee C, Clarke L, Auton A, Erlich Y, HandsakerRE, 1000 Genomes Project Consortium, Bustamante CD, Tyler-Smith C
Note: 'Punctuated bursts in human male demography inferred from 1,244 worldwide Y-chromosomesequences' by Poznik GD, Xue Y, Mendez FL, Willems TF, Massaia A, Wilson Sayres MA, Ayub Q,McCarthy SA, Narechania A, Kashin S, Chen Y, Banerjee R, Rodriguez-Flores JL, Cerezo M, ShaoH, Gymrek M, Malhotra A, Louzada S, Desalle R, Ritchie GR, Cerveira E, Fitzgerald TW, Garrison E,Marcketta A, Mittelman D, Romanovitch M, Zhang C, Zheng-Bradley X, Abecasis GR, McCarroll SA,Flicek P, Underhill PA, Coin L, Zerbino DR, Yang F, Lee C, Clarke L, Auton A, Erlich Y, HandsakerRE, 1000 Genomes Project Consortium, Bustamante CD, Tyler-Smith C. Nat Genet. 2016 Apr 25.doi: 10.1038/ng.3559.
Abstract: Protein–protein interactions (PPIs) play a vital role in most biological processes. Hence their comprehension can promote a better understanding of the mechanisms underlying living systems. However, besides the cost and the time limitation involved in the detection of experimentally validated PPIs, the noise in the data is still an important issue to overcome. In the last decade several in silico PPI prediction methods using both structural and genomic information were developed for this purpose. Here we introduce a unique validation approach aimed to collect reliable non interacting proteins (NIPs). Thereafter the most relevant protein/protein-pair related features were selected. Finally, the prepared dataset was used for PPI classification, leveraging the prediction capabilities of well-established machine learning methods. Our best classification procedure displayed specificity and sensitivity values of 96.33% and 98.02%, respectively, surpassing the prediction capabilities of other methods, including those trained on gold standard datasets. We showed that the PPI/NIP predictive performances can be considerably improved by focusing on data preparation.
Authors: Srivastava A, MazzoccoG, Kel A, Wyrwicz LS, Plewczynski D
Note: 'Detecting reliable non interacting proteins (NIPs) significantly enhancing the computational prediction of protein-protein interactions using machine learning methods' Srivastava A, MazzoccoG, Kel A, Wyrwicz LS, Plewczynski D. Mol Biosyst. 2016 Jan 7.
2015
Authors: PlewczynskiD, Gruca S, Szałaj P, Gulik K, de Oliveira SF, Malhotra A
Note: 'Analysis of Structural Chromosome Variants by Next Generation Sequencing Methods' PlewczynskiD, Gruca S, Szałaj P, Gulik K, de Oliveira SF and Malhotra A. book chapter in 'Clinical Applicationsfor Next-Generation Sequencing' book, Elsevier, 2015
Abstract: The 1000 Genomes Project set out to provide a comprehensive description of common human genetic variation by applying whole-genome sequencing to a diverse set of individuals from multiple populations. Here we report completion of the project, having reconstructed the genomes of 2,504 individuals from 26 populations using a combination of low-coverage whole-genome sequencing, deep exome sequencing, and dense microarray genotyping. We characterized a broad spectrum of genetic variation, in total over 88 million variants (84.7 million single nucleotide polymorphisms (SNPs), 3.6 million short insertions/deletions (indels), and 60,000 structural variants), all phased onto high-quality haplotypes. This resource includes >99% of SNP variants with a frequency of >1% for a variety of ancestries. We describe the distribution of genetic variation across the global sample, and discuss the implications for common disease studies.
Authors: 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR
Note: 'A global reference for human genetic variation' by 1000 Genomes Project Consortium,Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S,McVean GA, Abecasis GR. Nature. 2015 Oct 1;526(7571):68-74. doi: 10.1038/nature15393.
Abstract: Structural variants are implicated in numerous diseases and make up the majority of varying nucleotides among human genomes. Here we describe an integrated set of eight structural variant classes comprising both balanced and unbalanced variants, which we constructed using short-read DNA sequencing data and statistically phased onto haplotype blocks in 26 human populations. Analysing this set, we identify numerous gene-intersecting structural variants exhibiting population stratification and describe naturally occurring homozygous gene knockouts that suggest the dispensability of a variety of human genes. We demonstrate that structural variants are enriched on haplotypes identified by genome-wide association studies and exhibit enrichment for expression quantitative trait loci. Additionally, we uncover appreciable levels of structural variant complexity at different scales, including genic loci subject to clusters of repeated rearrangement and complex structural variants with multiple breakpoints likely to have formed through individual mutational events. Our catalogue will enhance future studies into structural variant demography, functional impact and disease association.
Authors: Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Hsi-Yang Fritz M, Konkel MK, Malhotra A, Stütz AM, Shi X, Paolo Casale F, Chen J, Hormozdiari F, Dayama G, ChenK, Malig M, Chaisson MJ, Walter K, Meiers S, Kashin S, Garrison E, Auton A, Lam HY, Jasmine MuX, Alkan C, Antaki D, Bae T, Cerveira E, Chines P, Chong Z, Clarke L, Dal E, Ding L, Emery S, FanX, Gujral M, Kahveci F, Kidd JM, Kong Y, Lameijer EW, McCarthy S, Flicek P, Gibbs RA, Marth G, Mason CE, Menelaou A, Muzny DM, Nelson BJ, Noor A, Parrish NF, Pendleton M, Quitadamo A, Raeder B, Schadt EE, Romanovitch M, Schlattl A, Sebra R, Shabalin AA, Untergasser A, Walker JA, Wang M, Yu F, Zhang C, Zhang J, Zheng-Bradley X, Zhou W, Zichner T, Sebat J, Batzer MA, McCarroll SA; 1000 Genomes Project Consortium, Mills RE, Gerstein MB, Bashir A, Stegle O, Devine SE, Lee C, Eichler EE, Korbel JO
Note: 'An integrated map of structural variation in 2,504 human genomes' by Sudmant PH, Rausch T,Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Hsi-Yang Fritz M,Konkel MK, Malhotra A, Stütz AM, Shi X, Paolo Casale F, Chen J, Hormozdiari F, Dayama G, ChenK, Malig M, Chaisson MJ, Walter K, Meiers S, Kashin S, Garrison E, Auton A, Lam HY, Jasmine MuX, Alkan C, Antaki D, Bae T, Cerveira E, Chines P, Chong Z, Clarke L, Dal E, Ding L, Emery S, FanX, Gujral M, Kahveci F, Kidd JM, Kong Y, Lameijer EW, McCarthy S, Flicek P, Gibbs RA, Marth G,Mason CE, Menelaou A, Muzny DM, Nelson BJ, Noor A, Parrish NF, Pendleton M, Quitadamo A,Raeder B, Schadt EE, Romanovitch M, Schlattl A, Sebra R, Shabalin AA, Untergasser A, Walker JA,Wang M, Yu F, Zhang C, Zhang J, Zheng-Bradley X, Zhou W, Zichner T, Sebat J, Batzer MA,McCarroll SA; 1000 Genomes Project Consortium, Mills RE, Gerstein MB, Bashir A, Stegle O,Devine SE, Lee C, Eichler EE, Korbel JO. Nature. 2015 Oct 1;526(7571):75-81. doi: 10.1038/nature15394.
Abstract: Glyceraldehyde-3-phosphate dehydrogenase from human sperm (GAPDHS) provides energy to the sperm flagellum, and is therefore essential for sperm motility and male fertility. This isoform is distinct from somatic GAPDH, not only in being specific for the testis but also because it contains an additional amino-terminal region that encodes a proline-rich motif that is known to bind to the fibrous sheath of the sperm tail. By conducting a large-scale sequence comparison on low-complexity sequences available in databases, we identified a strong similarity between the proline-rich motif from GAPDHS and the proline-rich sequence from Ena/vasodilator-stimulated phosphoprotein-like (EVL), which is known to bind an SH3 domain of dynamin-binding protein (DNMBP). The putative binding partners of the proline-rich GAPDHS motif include SH3 domain-binding protein 4 (SH3BP4) and the IL2-inducible T-cell kinase/tyrosine-protein kinase ITK/TSK (ITK). This result implies that GAPDHS participates in specific signal-transduction pathways. Gene Ontology category-enrichment analysis showed several functional classes shared by both proteins, of which the most interesting ones are related to signal transduction and regulation of hydrolysis. Furthermore, a mutation of one EVL proline to leucine is known to cause colorectal cancer, suggesting that mutation of homologous amino acid residue in the GAPDHS motif may be functionally deleterious.
Authors: Tatjewski M, Gruca A, Plewczynski D, Grynberg M
Note: 'The proline-rich region of glyceraldehyde-3-phosphate dehydrogenase from human sperm may bindSH3 domains, as revealed by a bioinformatic study of low-complexity protein segments' Tatjewski M,Gruca A, Plewczynski D, Grynberg M. Mol Reprod Dev. 2015 Dec 11. doi: 10.1002/mrd.22606.
2014
Abstract: The aftermath of influenza infection is determined by a complex set of host-pathogen interactions, where genomic variability on both viral and host sides influences the final outcome. Although there exists large body of literature describing influenza virus variability, only a very small fraction covers the issue of host variance. The goal of this review is to explore the variability of host genes responsible for host-pathogen interactions, paying particular attention to genes responsible for the presence of sialylated glycans in the host endothelial membrane, mucus, genes used by viral immune escape mechanisms, and genes particularly expressed after vaccination, since they are more likely to have a direct influence on the infection outcome.
Authors: Arcanjo AC, Mazzocco G, de Oliveira SF, Plewczynski D, Radomski JP
Note: 'Role of the host genetic variability in the influenza A virus susceptibility' by Arcanjo AC, Mazzocco G, de Oliveira SF, Plewczynski D, Radomski JP. Acta Biochim Pol. 2014; 61(3):403-19. Epub 2014 Sep 4.
BIOLOGICAL SYSTEMS MODELING
2016
Abstract: Here, we present two perspectives on the task of predicting post translational modifications (PTMs) from local sequence fragments using machine learning algorithms. The first is the description of the fundamental steps required to construct a PTM predictor from the very beginning. These steps include data gathering, feature extraction, or machine-learning classifier selection. The second part of our work contains the detailed discussion of more advanced problems which are encountered in PTM prediction task. Probably the most challenging issues which we have covered here are: (1) how to address the training data class imbalance problem (we also present statistics describing the problem); (2) how to properly set up cross-validation folds with an approach which takes into account the homology of protein data records, to address this problem we present our folds-over-clusters algorithm; and (3) how to efficiently reach for new sources of learning features. Presented techniques and notes resulted from intense studies in the field, performed by our and other groups, and can be useful both for researchers beginning in the field of PTM prediction and for those who want to extend the repertoire of their research techniques.
Authors: Tatjewski M, Kierczak M, Plewczynski D
Note: 'Predicting Post-Translational Modifications from Local Sequence Fragments Using Machine Learning Algorithms: Overview and Best Practices.' by Tatjewski M, Kierczak M, Plewczynski D. Methods Mol Biol. 2017;1484:275-300.
Abstract: The influenza virus type A (IVA) is an important pathogen which is able to cause annual epidemics and even pandemics. This fact is the consequence of the antigenic shifts and drifts capabilities of IVA, caused by the high mutation rate and the reassortment capabilities of the virus. The hemagglutinin (HA) protein constitutes the main IVA antigen and has a crucial role in the infection mechanism, being responsible for the recognition of host-specific sialic acid derivatives. Despite the relative abundance of HA sequence and serological studies, comparative structure-based analysis of HA are less investigated. The 3DFlu database contains well annotated HA representatives: 1192 models and 263 crystallographic structures. The relations between these proteins are defined using different metrics and are visualized as a network in the provided web interface. Moreover structural and sequence comparison of the proteins can be explored. Metadata information (e.g. protein identifier, IVA strain, year and location of infection) can enhance the exploration of the presented data. With our database researchers gain a useful tool for the exploration of high quality HA models, viewing and comparing changes in the HA viral subtypes at several information levels (sequence, structure, ESP). The complete and integrated view of those relations might be useful to determine the efficiency of transmission, pathogenicity and for the investigation of evolutionary tendencies of the influenza virus.Database URL: http://nucleus3d.cent.uw.edu.pl/influenza.
Authors: Mazzocco G, Lazniewski M, Migdał P, Szczepińska T, Radomski JP, Plewczynski D
Note: '3DFlu: database of sequence and structural variability of the influenza hemagglutinin at population scale.' by Mazzocco G, Lazniewski M, Migdał P, Szczepińska T, Radomski JP, Plewczynski D. Database (Oxford). 2016 Oct 2;2016. pii: baw130.
Abstract: Motivation: Accurate and effective dendritic spine segmentation from the dendrites remains as a challenge for current neuroimaging research community. In this paper, we present a new method (2dSpAn) for 2-d segmentation, classification and analysis of structural/plastic changes of hippocampal dendritic spines. A user interactive segmentation method with convolution kernels is designed to segment the spines from the dendrites. Formal morphological definitions are presented to describe key attributes related to the shape of segmented spines. Spines are automatically classified into one of four classes: Stubby, Filopodia, Mushroom and Spine-head Protrusions. Results: The developed method is validated using confocal light microscopy images of dendritic spines from dissociated hippocampal cultures for: 1) quantitative analysis of spine morphological changes, 2) reproducibility analysis for assessment of user-independence of the developed software, 3) accuracy analysis with respect to the manually labeled ground truth images, and also with respect to the available state-of-the-art. The developed method is monitored and used to precisely describe the morphology of individual spines in real-time experiments, i.e., consequent images of the same dendritic fragment. Availability: The software and the source code are available at https://sites.google.com/site/2dspan/ under open-source license for non-commercial use.
Authors: Subhadip Basu, Dariusz Plewczynski, Satadal Saha, MatyldaRoszkowska, Marta Magnowska, Ewa Baczynska and Jakub Wlodarczyk
Note: '2dSpAn: semiautomated 2-d segmentation, classification and analysis of hippocampaldendritic spine plasticity' by Subhadip Basu, Dariusz Plewczynski, Satadal Saha, MatyldaRoszkowska, Marta Magnowska, Ewa Baczynska and Jakub Wlodarczyk. Bioinformatics 2016 doi: 10.1093/bioinformatics/btw172 First published online: April 1, 2016
Abstract: ChIA-PET and Hi-C are high throughput versions of 3C-based mapping technologies that reveal long-range chromatin interactions and provide insights into the basic principles of spatial genome organization and gene regulation. Recently, we showed that a single ChIA-PET experiment provides information at all genomic scales of interest, from the high resolution locations of binding sites and enriched chromatin interactions mediated by specific protein factors, to the low resolution non-enriched interactions that reflect topological neighborhoods of higher-order associations. This multilevel nature of ChIA-PET data offers us an opportunity to use multiscale 3D models to study structural-functional relationships at multiple length scales, but doing so requires a structural modeling platform, which takes advantage of the full range of ChIA-PET data. Here we report 3D-NOME (3-Dimensional NucleOme Modeling Engine), a complete computational pipeline for processing and analyzing ChIA-PET data. 3D-NOME consists of three integrated tools: a graph-distance-based heatmap normalization tool, a 3D modeling platform, and an interactive 3D visualization tool. We use ChIA-PET and Hi-C data of human B-lymphocytes to demonstrate the effectiveness of 3D-NOME in building 3D genome models at multiple levels, including the entire nucleome, individual chromosomes, and specific segments at megabase (Mb) and kilobase (kb) resolutions. Our simulation protocol generates a single average structure or an ensemble of structures. We incorporate CTCF-motif orientation and high-resolution looping patterns in order to achieve more reliable, biologically plausible structures.
Authors: Szałaj P, Tang Z, Michalski P, Pietal MJ, Luo OJ, Sadowski M, Li X, Radew K, Ruan Y, Plewczynski D
Note: 'An integrated 3-dimensional genome modeling engine for data-driven simulation of spatial genome organization.' by Szałaj P, Tang Z, Michalski P, Pietal MJ, Luo OJ, Sadowski M, Li X, Radew K, Ruan Y, Plewczynski D. Genome Res. 2016 Oct 27. pii: gr.205062.116. [Epub ahead of print]
2015
Abstract: Accurate identification of protein–protein interactions (PPI) is the key step in understanding proteins’ biological functions, which are typically context-dependent. Many existing PPI predictors rely on aggregated features from protein sequences, however only a few methods exploit local information about specific residue contacts. In this work we present a two-stage machine learning approach for prediction of protein–protein interactions. We start with the carefully filtered data on protein complexes available for Saccharomyces cerevisiae in the Protein Data Bank (PDB) database. First, we build linear descriptions of interacting and non-interacting sequence segment pairs based on their inter-residue distances. Secondly, we train machine learning classifiers to predict binary segment interactions for any two short sequence fragments. The final prediction of the protein–protein interaction is done using the 2D matrix representation of all-against-all possible interacting sequence segments of both analysed proteins. The level-I predictor achieves 0.88 AUC for micro-scale, i.e., residue-level prediction. The level-II predictor improves the results further by a more complex learning paradigm. We perform 30-fold macro-scale, i.e., protein-level cross-validation experiment. The level-II predictor using PSIPRED-predicted secondary structure reaches 0.70 precision, 0.68 recall, and 0.70 AUC, whereas other popular methods provide results below 0.6 threshold (recall, precision, AUC). Our results demonstrate that multi-scale sequence features aggregation procedure is able to improve the machine learning results by more than 10% as compared to other sequence representations. Prepared datasets and source code for our experimental pipeline are freely available for download from: http://zubekj.github.io/mlppi/ (open source Python implementation, OS independent).
Authors: Zubek J, Tatjewski M, Boniecki A, Mnich M, Basu S, Plewczynski D
Note: 'Multi-level machine learning prediction of protein-protein interactions in Saccharomyces cerevisiae'by Zubek J, Tatjewski M, Boniecki A, Mnich M, Basu S, Plewczynski D. PeerJ. 2015 Jul 2;3:e1041.doi: 10.7717/peerj.1041.
Abstract: Bacteria are increasingly resistant to existing antibiotics, which target a narrow range of pathways. New methods are needed to identify targets, including repositioning targets among distantly related species. We developed a novel combination of systems and structural modeling and bioinformatics to reposition known antibiotics and targets to new species. We applied this approach to Mycoplasma genitalium, a common cause of urethritis. First, we used quantitative metabolic modeling to identify enzymes whose expression affects the cellular growth rate. Second, we searched the literature for inhibitors of homologs of the most fragile enzymes. Next, we used sequence alignment to assess that the binding site is shared by M. genitalium, but not by humans. Lastly, we used molecular docking to verify that the reported inhibitors preferentially interact with M. genitalium proteins over their human homologs. Thymidylate kinase was the top predicted target and piperidinylthymines were the top compounds. Further work is needed to experimentally validate piperidinylthymines. In summary, combined systems and structural modeling is a powerful tool for drug repositioning.
Authors: Kazakiewicz D, Karr JR, Langner KM, Plewczynski D
Note: 'A combined systems and structural modeling approach repositions antibiotics for Mycoplasma genitalium' by Kazakiewicz D, Karr JR, Langner KM, Plewczynski D. Comput Biol Chem. 2015 Jul 30. pii: S1476-9271(15)30089-X. doi: 10.1016/j.compbiolchem.2015.07.007.
Abstract: Whole-cell models that explicitly represent all cellular components at the molecular level have the potential to predict phenotype from genotype. However, even for simple bacteria, whole-cell models will contain thousands of parameters, many of which are poorly characterized or unknown. New algorithms are needed to estimate these parameters and enable researchers to build increasingly comprehensive models. We organized the Dialogue for Reverse Engineering Assessments and Methods (DREAM) 8 Whole-Cell Parameter Estimation Challenge to develop new parameter estimation algorithms for whole-cell models. We asked participants to identify a subset of parameters of a whole-cell model given the model’s structure and in silico “experimental” data. Here we describe the challenge, the best performing methods, and new insights into the identifiability of whole-cell models. We also describe several valuable lessons we learned toward improving future challenges. Going forward, we believe that collaborative efforts supported by inexpensive cloud computing have the potential to solve whole-cell model parameter estimation.
Authors: Karr JR, Williams AH, Zucker JD, Raue A, Steiert B, Timmer J, Kreutz C, DREAM8 Parameter Estimation Challenge Consortium, Wilkinson S, Allgood BA, Bot BM, Hoff BR, Kellen MR, Covert MW, Stolovitzky GA, Meyer P
Note: 'Summary of the DREAM8 Parameter Estimation Challenge: Toward Parameter Identification forWhole-Cell Models' by Karr JR, Williams AH, Zucker JD, Raue A, Steiert B, Timmer J, Kreutz C;DREAM8 Parameter Estimation Challenge Consortium, Wilkinson S, Allgood BA, Bot BM, Hoff BR,Kellen MR, Covert MW, Stolovitzky GA, Meyer P. PLoS Comput Biol. 2015 May 28;11(5):e1004096.doi: 10.1371/journal.pcbi.1004096.
Abstract: Motivation: To date, only a few distinct successful approaches have been introduced to reconstruct a protein 3D structure from a map of contacts between its amino acid residues (a 2D contact map). Current algorithms can infer structures from information-rich contact maps that contain a limited fraction of erroneous predictions. However, it is difficult to reconstruct 3D structures from predicted contact maps that usually contain a high fraction of false contacts. Results: We describe a new, multi-step protocol that predicts protein 3D structures from the predicted contact maps. The method is based on a novel distance function acting on a fuzzy residue proximity graph, which predicts a 2D distance map from a 2D predicted contact map. The application of a Multi-Dimensional Scaling algorithm transforms that predicted 2D distance map into a coarse 3D model, which is further refined by typical modeling programs into an all-atom representation. We tested our approach on contact maps predicted de novo by MULTICOM, the top contact map predictor according to CASP10. We show that our method outperforms FT-COMAR, the state-of-the-art method for 3D structure reconstruction from 2D maps. For all predicted 2D contact maps of relatively low sensitivity (60–84%), GDFuzz3D generates more accurate 3D models, with the average improvement of 4.87 Å in terms of RMSD. Availability: GDFuzz3D server and standalone version are freely available at http://iimcb.genesilico.pl/gdserver/GDFuzz3D/.
Authors: Michal J Pietal, Janusz M Bujnicki, and Lukasz P Kozlowski
Note: 'GDFuzz3D: a method for protein 3D structure reconstruction from contact maps, based on a non-Euclidean distance function' by Michal J. Pietal, Janusz M. Bujnicki, and Lukasz P. Kozlowski. Bioinformatics 2015 doi: 10.1093/bioinformatics/btv390 First published online: June 30, 2015
2013
Abstract: Class II human leukocyte antigens (HLA II) are proteins involved in the human immunological adaptive response by binding and exposing some pre-processed, non-self peptides in the extracellular domain in order to make them recognizable by the CD4+ T lymphocytes. However, the understanding of HLA-peptide binding interaction is a crucial step for designing a peptide-based vaccine because the high rate of polymorphisms in HLA class II molecules creates a big challenge, even though the HLA II proteins can be grouped into supertypes, where members of different class bind a similar pool of peptides. Hence, first we performed the supertype classification of 27 HLA II proteins using their binding affinities and structural-based linear motifs to create a stable group of supertypes. For this purpose, a well-known clustering method was used, and then, a consensus was built to find the stable groups and to show the functional and structural correlation of HLA II proteins. Thus, the overlap of the binding events was measured, confirming a large promiscuity within the HLA II-peptide interactions. Moreover, a very low rate of locus-specific binding events was observed for the HLA-DP genetic locus, suggesting a different binding selectivity of these proteins with respect to HLA-DR and HLA-DQ proteins. Secondly, a predictor based on a support vector machine (SVM) classifier was designed to recognize HLA II-binding peptides. The efficiency of prediction was estimated using precision, recall (sensitivity), specificity, accuracy, F-measure, and area under the ROC curve values of random subsampled dataset in comparison with other supervised classifiers. Also the leave-one-out cross-validation was performed to establish the efficiency of the predictor. The availability of HLA II-peptide interaction dataset, HLA II-binding motifs, high-quality amino acid indices, peptide dataset for SVM training, and MATLAB code of the predictor is available at http://sysbio.icm.edu.pl/HLA .
Authors: I Saha, G Mazzocco and D Plewczynski
Note: 'Consensus classification of Human Leukocyte Antigens class II proteins' by I. Saha, G. Mazzocco and D. Plewczynski. Immunogenetics 65(2):97-105 (2013).
2011
Abstract: Protein-protein interactions (PPI) control most of the biological processes in a living cell. In order to fully understand protein functions, a knowledge of protein-protein interactions is necessary. Prediction of PPI is challenging, especially when the three-dimensional structure of interacting partners is not known. Recently, a novel prediction method was proposed by exploiting physical interactions of constituent domains. We propose here a novel knowledge-based prediction method, namely PPI_SVM, which predicts interactions between two protein sequences by exploiting their domain information. We trained a two-class support vector machine on the benchmarking set of pairs of interacting proteins extracted from the Database of Interacting Proteins (DIP). The method considers all possible combinations of constituent domains between two protein sequences, unlike most of the existing approaches. Moreover, it deals with both single-domain proteins and multi domain proteins; therefore it can be applied to the whole proteome in high-throughput studies. Our machine learning classifier, following a brainstorming approach, achieves accuracy of 86%, with specificity of 95%, and sensitivity of 75%, which are better results than most previous methods that sacrifice recall values in order to boost the overall precision. Our method has on average better sensitivity combined with good selectivity on the benchmarking dataset. The PPI_SVM source code, train/test datasets and supplementary files are available freely in the public domain at: http://code.google.com/p/cmater-bioinfo/.
Authors: P Chatterjee, S Basu, M Kundu, M Nasipuri, and D Plewczynski
Note: 'PPI_SVM: prediction of protein-protein interactions using machine learning, domain-domain affinities and frequency tables' by P. Chatterjee, S. Basu, M. Kundu, M. Nasipuri, and D. Plewczynski Cell Mol Biol Lett 16(2):264-78 (2011).
Abstract: Studying the interactome is one of the exciting frontiers of proteomics, as shown lately at the recent bioinformatics conferences (for example ISMB 2010, or ECCB 2010). Distribution of data is facilitated by a large number of databases. Metamining databases have been created in order to allow researchers access to several databases in one search, but there are serious difficulties for end users to evaluate the metamining effort. Therefore we suggest a new standard, 'Good Interaction Data Metamining Practice' (GIDMP), which could be easily automated and requires only very minor inclusion of statistical data on each database homepage. Widespread adoption of the GIDMP standard would provide users with: a standardized way to evaluate the statistics provided by each metamining database, thus enhancing the end-user experience; a stable contact point for each database, allowing the smooth transition of statistics; a fully automated system, enhancing time- and cost-effectiveness. The proposed information can be presented as a few hidden lines of text on the source database www page, and a constantly updated table for a metamining database included in the source/credits web page.
Authors: D Plewczynski, and T Klingström Cell
Note: 'GIDMP: good protein-protein interaction data metamining practice' by D. Plewczynski, and T. Klingström Cell Mol Biol Lett. 16(2):258-63 (2011).
BIOSTATISTICS AND COGNITIVE COMPUTING
2015
Abstract: With the avalanche of genomic and proteomic data generated in the postgenomic age, it is highly desirable to develop automated methods for rapidly and effectively analyzing and predicting the structure, function, and other properties of DNA and protein. Researchers realize the importance of machine learning methods and feature selection algorithms for potential knowledge finding tasks in genomics and proteomics. Recent years have shown tremendous advances in the properties prediction of DNA fragments and protein sequences by various pattern recognition methods. These techniques provide economical and time-saving solutions for identifying the properties of DNA and protein. This special issue will focus on various aspects of the application of machine learning methods in genomics and proteomics bioinformatics. The recent developments on the prediction of protein subcellular localization, posttranslational modification sites, DNA-binding site, protein-protein interaction, nucleosome positioning, transcription factor binding site, exon/intron splice site, translation initiation site, and transcription start site will be included in the special issue.
Authors: Lin H, Chen W, Anandakrishnan R, Plewczynski D
Note: 'Application of machine learning method in genomics and proteomics' by Lin H, Chen W,Anandakrishnan R, Plewczynski D. Scientific World Journal. 2015; 2015:914780. doi:10.1155/2015/914780. Epub 2015 Apr 19.
Abstract: The Cyclin-Dependent Kinases (CDKs) are the core components coordinating eukaryotic cell division cycle. Generally the crystal structure of CDKs provides information on possible molecular mechanisms of ligand binding. However, reliable and robust estimation of ligand binding activity has been a challenging task in drug design. In this regard, various machine learning techniques, such as Support Vector Machine, Naive Bayesian classifier, Decision Tree, and K-Nearest Neighbor classifier, have been used. The performance of these heterogeneous classification techniques depends on proper selection of features from the data set. This fact motivated us to propose an integrated classification technique using Genetic Algorithm (GA), Rotational Feature Selection (RFS) scheme, and Ensemble of Machine Learning methods, named as the Genetic Algorithm integrated Rotational Ensemble based classification technique, for the prediction of ligand binding activity of CDKs. This technique can automatically find the important features and the ensemble size. For this purpose, GA encodes the features and ensemble size in a chromosome as a binary string. Such encoded features are then used to create diverse sets of training points using RFS in order to train the machine learning method multiple times. The RFS scheme works on Principal Component Analysis (PCA) to preserve the variability information of the rotational nonoverlapping subsets of original data. Thereafter, the testing points are fed to the different instances of trained machine learning method in order to produce the ensemble result. Here accuracy is computed as a final result after 10-fold cross validation, which also used as an objective function for GA to maximize. The effectiveness of the proposed classification technique has been demonstrated quantitatively and visually in comparison with different machine learning methods for 16 ligand binding CDK docking and rescoring data sets. In addition, the best possible features have been reported for CDK docking and rescoring data sets separately. Finally, the Friedman test has been conducted to judge the statistical significance of the results produced by the proposed technique. The results indicate that the integrated classification technique has high relevance in predicting of protein-ligand binding activity.
Authors: Saha I, Rak B, Bhowmick SS, Maulik U, Bhattacharjee D, Koch U, Lazniewski M, Plewczynski D
Note: 'Binding Activity Prediction of Cyclin-Dependent Inhibitors' by Saha I, Rak B, Bhowmick SS,Maulik U, Bhattacharjee D, Koch U, Lazniewski M, Plewczynski D. J Chem Inf Model. 2015 Jul27;55(7):1469-82. doi: 10.1021/ci500633c.
2014
Abstract: Protein–protein interactions are important for the majority of biological processes. A significant number of computational methods have been developed to predict protein–protein interactions using protein sequence, structural and genomic data. Vast experimental data is publicly available on the Internet, but it is scattered across numerous databases. This fact motivated us to create and evaluate new high-throughput datasets of interacting proteins. We extracted interaction data from DIP, MINT, BioGRID and IntAct databases. Then we constructed descriptive features for machine learning purposes based on data from Gene Ontology and DOMINE. Thereafter, four well-established machine learning methods: Support Vector Machine, Random Forest, Decision Tree and Naïve Bayes, were used on these datasets to build an Ensemble Learning method based on majority voting. In cross-validation experiment, sensitivity exceeded 80% and classification/prediction accuracy reached 90% for the Ensemble Learning method. We extended the experiment to a bigger and more realistic dataset maintaining sensitivity over 70%. These results confirmed that our datasets are suitable for performing PPI prediction and Ensemble Learning method is well suited for this task. Both the processed PPI datasets and the software are available at http://sysbio.icm.edu.pl/indra/EL-PPI/home.html.
Authors: Saha I, Zubek J, Klingström T, Forsberg S, Wikander J, Kierczak M, Maulik U, Plewczynski D
Note: 'Ensemble learning prediction of protein-protein interactions using proteins functional annotations' by Saha I, Zubek J, Klingström T, Forsberg S, Wikander J, Kierczak M, Maulik U, Plewczynski D. Mol Biosyst. 2014 Apr;10(4):820-30. doi: 10.1039/c3mb70486f.
2013
Abstract: We study mathematical models of the collaborative solving of a two-choice discrimination task. We estimate the difference between the shared performance for a group of nn observers over a single person performance. Our paper is a theoretical extension of the recent work of Bahrami, Olsen, Latham, Roepstorff, and Frith (2010) from a dyad (a pair) to a group of nn interacting minds. We analyze several models of communication, decision-making and hierarchical information-aggregation. The maximal slope of psychometric function is a convenient parameter characterizing performance. For every model we investigated, the group performance turns out to be a product of two numbers: a scaling factor depending of the group size and an average performance. The scaling factor is a power function of the group size (with the exponent ranging from 0 to 1), whereas the average is arithmetic mean, quadratic mean, or maximum of the individual slopes. Moreover, voting can be almost as efficient as more elaborate communication models, given the participants have similar individual performances.
Authors: P Migdał, J Rączaszek- Leonardi, M Denkiewicz and D Plewczynski
Note: 'Information-sharing and aggregation models for interacting minds' by P. Migdał, J. Rączaszek- Leonardi, M. Denkiewicz and D. Plewczynski. Journal of Mathematical Psychology 56: 417-426 (2013).
Abstract: The physico-chemical properties of interaction interfaces have a crucial role in characterization of protein-protein interactions (PPI). In silico prediction of participating amino acids helps to identify interface residues for further experimental verification using mutational analysis, or inhibition studies by screening library of ligands against given protein. Given the unbound structure of a protein and the fact that it forms a complex with another known protein, the objective of this work is to identify the residues that are involved in the interaction. We attempt to predict interaction sites in protein complexes using local composition of amino acids together with their physico-chemical characteristics. The local sequence segments (LSS) are dissected from the protein sequences using a sliding window of 21 amino acids. The list of LSSs is passed to the support vector machine (SVM) predictor, which identifies interacting residue pairs considering their inter-atom distances. We have analyzed three different model organisms of Escherichia coli, Saccharomyces Cerevisiae and Homo sapiens, where the numbers of considered hetero-complexes are equal to 40, 123 and 33 respectively. Moreover, the unified multi-organism PPI meta-predictor is also developed under the current work by combining the training databases of above organisms. The PPIcons interface residues prediction method is measured by the area under ROC curve (AUC) equal to 0.82, 0.75, 0.72 and 0.76 for the aforementioned organisms and the meta-predictor respectively.
Authors: BK Sriwastava, S Basu, U Maulik, D Plewczynski
Note: 'PPIcons: identification of protein-protein interaction sites in selected organisms' by BK. Sriwastava, S. Basu, U. Maulik, D. Plewczynski. J Mol Model 19(9):4059-70 (2013).
2012
Abstract: We present here the 2011 update of the AutoMotif Service (AMS 4.0) that predicts the wide selection of 88 different types of the single amino acid post-translational modifications (PTM) in protein sequences. The selection of experimentally confirmed modifications is acquired from the latest UniProt and Phospho.ELM databases for training. The sequence vicinity of each modified residue is represented using amino acids physico-chemical features encoded using high quality indices (HQI) obtaining by automatic clustering of known indices extracted from AAindex database. For each type of the numerical representation, the method builds the ensemble of Multi-Layer Perceptron (MLP) pattern classifiers, each optimising different objectives during the training (for example the recall, precision or area under the ROC curve (AUC)). The consensus is built using brainstorming technology, which combines multi-objective instances of machine learning algorithm, and the data fusion of different training objects representations, in order to boost the overall prediction accuracy of conserved short sequence motifs. The performance of AMS 4.0 is compared with the accuracy of previous versions, which were constructed using single machine learning methods (artificial neural networks, support vector machine). Our software improves the average AUC score of the earlier version by close to 7 % as calculated on the test datasets of all 88 PTM types. Moreover, for the selected most-difficult sequence motifs types it is able to improve the prediction performance by almost 32 %, when compared with previously used single machine learning methods. Summarising, the brainstorming consensus meta-learning methodology on the average boosts the AUC score up to around 89 %, averaged over all 88 PTM types. Detailed results for single machine learning methods and the consensus methodology are also provided, together with the comparison to previously published methods and state-of-the-art software tools. The source code and precompiled binaries of brainstorming tool are available at http://code.google.com/p/automotifserver/ under Apache 2.0 licensing.
Authors: D Plewczynski, S Basu and I Saha
Note: 'AMS 4.0: consensus prediction of post-translational modifications in protein sequences' by D. Plewczynski, S. Basu and I. Saha. Amino Acids 43(2):573-82 (2012).
Abstract: In this article, we categorize presently available experimental and theoretical knowledge of various physicochemical and biochemical features of amino acids, as collected in the AAindex database of known 544 amino acid (AA) indices. Previously reported 402 indices were categorized into six groups using hierarchical clustering technique and 142 were left unclustered. However, due to the increasing diversity of the database these indices are overlapping, therefore crisp clustering method may not provide optimal results. Moreover, in various large-scale bioinformatics analyses of whole proteomes, the proper selection of amino acid indices representing their biological significance is crucial for efficient and error-prone encoding of the short functional sequence motifs. In most cases, researchers perform exhaustive manual selection of the most informative indices. These two facts motivated us to analyse the widely used AA indices. The main goal of this article is twofold. First, we present a novel method of partitioning the bioinformatics data using consensus fuzzy clustering, where the recently proposed fuzzy clustering techniques are exploited. Second, we prepare three high quality subsets of all available indices. Superiority of the consensus fuzzy clustering method is demonstrated quantitatively, visually and statistically by comparing it with the previously proposed hierarchical clustered results. The processed AAindex1 database, supplementary material and the software are available at http://sysbio.icm.edu.pl/aaindex/ .
Authors: I Saha, U Maulik, S Bandyopadhyay and D Plewczynski
Note: 'Fuzzy Clustering of Physicochemical and Biochemical Properties of Amino Acids' by I. Saha, U. Maulik, S. Bandyopadhyay and D. Plewczynski. Amino Acids 43(2):583-94 (2012).
Abstract: Clustering is an important tool for analysing the microarray data to identify groups of co-expressed genes. The problem of fuzzy clustering in microarray data motivated us to develop an improved clustering algorithm. In this paper, an improved differential evolution based fuzzy clustering technique is proposed. The performance of the proposed improved differential evolution based fuzzy clustering technique has been compared with other state-of-the-art clustering algorithms for publicly available benchmark microarray data sets. Statistical and biological significance tests have been carried out to establish the statistical superiority of the proposed clustering approach and biological relevance of clusters of co-expressed genes, respectively.
Authors: I Saha, D Plewczynski, U Maulik and S Bandyopadhyay
Note: 'Improved differential evolution for microarray analysis' by I. Saha, D. Plewczynski, U. Maulik and S. Bandyopadhyay. J. Data Mining and Bioinformatics 6(1):86-103 (2012).
2011
Abstract: Secondary structure prediction is a crucial task for understanding the variety of protein structures and performed biological functions. Prediction of secondary structures for new proteins using their amino acid sequences is of fundamental importance in bioinformatics. We propose a novel technique to predict protein secondary structures based on position-specific scoring matrices (PSSMs) and physico-chemical properties of amino acids. It is a two stage approach involving multiclass support vector machines (SVMs) as classifiers for three different structural conformations, viz., helix, sheet and coil. In the first stage, PSSMs obtained from PSI-BLAST and five specially selected physicochemical properties of amino acids are fed into SVMs as features for sequence-to-structure prediction. Confidence values for forming helix, sheet and coil that are obtained from the first stage SVM are then used in the second stage SVM for performing structure-to-structure prediction. The two-stage cascaded classifiers (PSP_MCSVM) are trained with proteins from RS126 dataset. The classifiers are finally tested on target proteins of critical assessment of protein structure prediction experiment-9 (CASP9). PSP_MCSVM with brainstorming consensus procedure performs better than the prediction servers like Predator, DSC, SIMPA96, for randomly selected proteins from CASP9 targets. The overall performance is found to be comparable with the current state-of-the art. PSP_MCSVM source code, train-test datasets and supplementary files are available freely in public domain at: http://sysbio.icm.edu.pl/secstruct and http://code.google.com/p/cmater-bioinfo/
Authors: P Chatterjee, S Basu, M Kundu, M Nasipuri, and D Plewczynski
Note: 'PSP_MCSVM: brainstorming consensus prediction of protein secondary structures using two-stage multiclass support vector machines' by P. Chatterjee, S. Basu, M. Kundu, M. Nasipuri, and D. Plewczynski. J Mol Model. 17(9):2191-201 (2011).
PHYSICAL CHEMISTRY
2002
Abstract: We focus on classical chaotic systems corrupted by white and colored noise. We study the dependence of the correlation dimension and the Kolmogorov entropy on the noise level and its spectral exponent. As is well known, white noise strongly reduces the width of the scaling region for the correlation dimension and entropy. On the contrary, we demonstrate that colored noise does not basically obscure the scaling region, changing only the shape of the correlation sum for length scales smaller than the noise level. The numerical results show that, even for a noise level as high as approximately 5%, a reasonably wide plateau for the correlation sum is still obtained, but the value of the calculated dimension is somewhat increased. The calculated correlation dimension is a bilinear function of the noise level and the dimension of the noise, which depends on the spectral exponent of the noise. On the other hand, the width of the scaling region for the correlation entropy depends on this spectral exponent, but the value of the plateau does not change substantially.
Authors: S Redaelli, D Plewczynski, W Macek
Note: 'Influence of Colored Noise on Chaotic Systems' by S. Redaelli, D. Plewczynski, W. Macek. Physical Review E 66, p. 035202 (2002);
2001
Abstract: We present a simple numerical analysis of the diffusion on a curved surface given by the equation Full-size image (<1 K) in a finite domain Full-size image (<1 K). The first non-vanishing eigenvalue of the Beltrami–Laplace operator with the reflecting boundary conditions is determined in our simulations for the P, D, G, S, S1 and I-WP, nodal periodic surfaces, where Full-size image (<1 K) is their respective cubic unit cell. We observe that the first eigenvalue for the surfaces of simple topology (P,D,G,I-WP) is smaller than for the surfaces of complex topology (S,S1).
Authors: D Plewczynski, R Holyst
Note: 'Approach to equilibrium of particles diffusing on curved surfaces' by D. Plewczynski, R. Holyst. Physica A 295, p. 371-378 (2001);
2000
Abstract: We present a catalogue of diffusion coefficients and reorientational angle distribution (RAD) for various periodic surfaces, such as I-WP, F-RD, S, and S1 nodal surfaces; cylindrical structures like simple, undulated, and spiral cylinders, and a three-dimensional interconnected-rod structures. The results are obtained on the basis of a simulation algorithm for a diffusion on a surface given by the general equation φ(r)=0 [Hołyst et al., Phys Rev. E 60, 302 (1999)]. I-WP, S, and S1 surfaces have a spherelike RAD, while F-RD has a cubelike RAD. The average of the second Legendre polynomial with RAD function for all nodal surfaces, except the F-RD nodal surface, decays exponentially with time for short times. The decay time is related to the Euler characteristic and the area per unit cell of a surface. This analytical formula, first proposed by B. Halle, S. Ljunggren, and S. Lidin in J. Chem. Phys. 97, 1401 (1992), is checked here on nodal surfaces, and its range of validity is determined. RAD function approaches its stationary limit exponentially with time. We determine the time to reach stationary state for all surfaces. In the case of the value of the effective diffusion coefficient the mean curvature and a connectivity between parts of surfaces have the main influence on it. The surfaces with low mean curvature at every point of the surface are characterized by high-diffusion coefficient. However if a surface has globally low mean curvature with large regions of nonzero mean curvature (negative and positive) the effective diffusion coefficient is low, as for example, in the case of undulated cylinders. Increasing the connectivity, at fixed curvatures, increases the diffusion coefficient.
Authors: D Plewczynski, R Holyst
Note: 'Reorientational angle distribution and diffusion coefficient for nodal and cylindrical surfaces' by D. Plewczynski, R. Holyst. J. Chem. Phys. 22, 9920 (2000);
1999
Abstract: We present a simulation algorithm for a diffusion on a curved surface given by the equation φ(r)=0. The algorithm is tested against analytical results known for diffusion on a cylinder and a sphere, and applied to the diffusion on the P, D, and G periodic nodal surfaces. It should find application in an interpretation of two-dimensional exchange NMR spectroscopy data of diffusion on biological membranes.
Authors: D Plewczynski, R Holyst, A Aksimientiev, K Burdzy
Note: 'Diffusion on Curved Surfaces' by D. Plewczynski, R. Holyst, A. Aksimientiev, K. Burdzy. Phys. Rev. E 60, p. 302 (1999);
1998
Abstract: We discuss the Landau theory for a nonlinear equation that can describe social changes, such as an influence of the social environment on individual. The models can explain why the minority can survive inside the majority population. It is described in terms of the complex intermittent clusters behavior in the stationary limit.
Authors: D Plewczynski
Note: 'Landau Theory of Social Clustering' by D. Plewczynski. Physica A 261, p. 608 (1998);
BIOINFORMATICS
2011
Abstract: Docking is one of the most commonly used techniques in drug design. It is used for both identifying correct poses of a ligand in the binding site of a protein as well as for the estimation of the strength of protein–ligand interaction. Because millions of compounds must be screened, before a suitable target for biological testing can be identified, all calculations should be done in a reasonable time frame. Thus, all programs currently in use exploit empirically based algorithms, avoiding systematic search of the conformational space. Similarly, the scoring is done using simple equations, which makes it possible to speed up the entire process. Therefore, docking results have to be verified by subsequent in vitro studies. The purpose of our work was to evaluate seven popular docking programs (Surflex, LigandFit, Glide, GOLD, FlexX, eHiTS, and AutoDock) on the extensive dataset composed of 1300 protein–ligands complexes from PDBbind 2007 database, where experimentally measured binding affinity values were also available. We compared independently the ability of proper posing [according to Root mean square deviation (or Root mean square distance) of predicted conformations versus the corresponding native one] and scoring (by calculating the correlation between docking score and ligand binding strength). To our knowledge, it is the first large-scale docking evaluation that covers both aspects of docking programs, that is, predicting ligand conformation and calculating the strength of its binding. More than 1000 protein–ligand pairs cover a wide range of different protein families and inhibitor classes. Our results clearly showed that the ligand binding conformation could be identified in most cases by using the existing software, yet we still observed the lack of universal scoring function for all types of molecules and protein families. © 2010 Wiley Periodicals, Inc. J Comput Chem, 2011
Authors: D Plewczynski, M Łaźniewski, R Augustyniak, and K Ginalski Journal
Note: 'Can we trust docking results? Evaluation of seven commonly used programs on PDBbind database' by D. Plewczynski, M. Łaźniewski, R. Augustyniak, and K. Ginalski. Journal Computational Chemistry 32(4):742-55. (2011);
Abstract: Molecular recognition plays a fundamental role in all biological processes, and that is why great efforts have been made to understand and predict protein-ligand interactions. Finding a molecule that can potentially bind to a target protein is particularly essential in drug discovery and still remains an expensive and time-consuming task. In silico, tools are frequently used to screen molecular libraries to identify new lead compounds, and if protein structure is known, various protein-ligand docking programs can be used. The aim of docking procedure is to predict correct poses of ligand in the binding site of the protein as well as to score them according to the strength of interaction in a reasonable time frame. The purpose of our studies was to present the novel consensus approach to predict both protein-ligand complex structure and its corresponding binding affinity. Our method used as the input the results from seven docking programs (Surflex, LigandFit, Glide, GOLD, FlexX, eHiTS, and AutoDock) that are widely used for docking of ligands. We evaluated it on the extensive benchmark dataset of 1300 protein-ligands pairs from refined PDBbind database for which the structural and affinity data was available. We compared independently its ability of proper scoring and posing to the previously proposed methods. In most cases, our method is able to dock properly approximately 20% of pairs more than docking methods on average, and over 10% of pairs more than the best single program. The RMSD value of the predicted complex conformation versus its native one is reduced by a factor of 0.5 Å. Finally, we were able to increase the Pearson correlation of the predicted binding affinity in comparison with the experimental value up to 0.5.
Authors: D Plewczynski, M Łaźniewski, M von Grotthuss, L Rychlewski and K Ginalski
Note: 'VoteDock: consensus docking method for prediction of protein-ligand interactions' by D. Plewczynski, M. Łaźniewski, M. von Grotthuss, L. Rychlewski and K. Ginalski. Journal Computational Chemistry 32(4):568-81 (2011);
Abstract: In this article, a novel concept is introduced by using both unsupervised and supervised learning. For unsupervised learning, the problem of fuzzy clustering in microarray data as a multiobjective optimization is used, which simultaneously optimizes two internal fuzzy cluster validity indices to yield a set of Pareto-optimal clustering solutions. In this regards, a new multiobjective differential evolution based fuzzy clustering technique has been proposed. Subsequently, for supervised learning, a fuzzy majority voting scheme along with support vector machine is used to integrate the clustering information from all the solutions in the resultant Pareto-optimal set. The performances of the proposed clustering techniques have been demonstrated on five publicly available benchmark microarray data sets. A detail comparison has been carried out with multiobjective genetic algorithm based fuzzy clustering, multiobjective differential evolution based fuzzy clustering, single objective versions of differential evolution and genetic algorithm based fuzzy clustering as well as well known fuzzy c-means algorithm. While using support vector machine, comparative studies of the use of four different kernel functions are also reported. Statistical significance test has been done to establish the statistical superiority of the proposed multiobjective clustering approach. Finally, biological significance test has been carried out using a web based gene annotation tool to show that the proposed integrated technique is able to produce biologically relevant clusters of coexpressed genes.
Authors: I Saha, U Maulik, S Bandyopadhyay and D Plewczynski
Note: 'Unsupervised and Supervised learning approaches together for Microarray Analysis' by I. Saha, U. Maulik, S. Bandyopadhyay and D. Plewczynski. Fundamenta Informaticae FUND INFORM 106 (1): 45-73 (2011);
Abstract: The amount of information regarding protein–protein interactions (PPI) at a proteomic scale is constantly increasing. This is paralleled with an increase of databases making information available. Consequently there are diverse ways of delivering information about not only PPIs but also regarding the databases themselves. This creates a time consuming obstacle for many researchers working in the field. Our survey provides a valuable tool for researchers to reduce the time necessary to gain a broad overview of PPI-databases and is supported by a graphical representation of data exchange. The graphical representation is made available in cooperation with the team maintaining www.pathguide.org and can be accessed at http://www.pathguide.org/interactions.php in a new Cytoscape web implementation. The local copy of Cytoscape cys file can be downloaded from http://bio.icm.edu.pl/∼darman/ppi web page.
Authors: T Klingström and D Plewczynski
Note: 'Protein-protein interaction and pathway databases, a graphical review' by T. Klingström and D. Plewczynski. Briefings in Bioinformatics 12(6):702-13 (2011);
Abstract: Squalene epoxidase (SE) is a key flavin adenine dinucleotide (FAD)-dependent enzyme of ergosterol and cholesterol biosynthetic pathways and an attractive potential target for drugs used to inhibit the growth of pathogenic fungi or to lower cholesterol level. Although many studies on allylamine drugs activity have been published during the last 30 years, up until now no detailed mechanism of the squalene epoxidase inhibition has been presented. Our study brings such a model at atomic resolution in the case of yeast Saccharomyces cerevisiae . Presented data resulting from modeling studies are in excellent agreement with experimental findings. A fully atomic three-dimensional (3D) model of squalene epoxidase (EC 1.14.99.7) from S. cerevisiae was built with the help of 3D-Jury approach and further screened based on data known from mutation experiments leading to terbinafine resistance. Docking studies followed by molecular dynamics simulations and quantum interaction energy calculations [MP2/6-31G(d)] resulted in the identification of the terbinafine-squalene epoxidase mode of interaction. In the energetically most likely orientation of terbinafine its interaction energy with the protein is ca. 120 kJ/mol. In the favorable position the terbinafine lipophilic moiety is located vertically inside the squalene epoxidase binding pocket with the tert-butyl group oriented toward its center. Such a position results in the SE conformational changes and prevents the natural substrate from being able to bind to the enzyme's active site. That would explain the noncompetitive manner of SE inhibition. We found that the strongest interaction between terbinafine and SE stems from hydrogen bonding between hydrogen-bond donors, hydroxyl group of Tyr90 and amine nitrogen atom of terbinafine. Moreover, strong attractive interactions were recorded for amino acids whose mutations resulted in terbinafine resistance. Our results, elucidating at a molecular level the mode of terbinafine inhibitory activity, can be utilized in designing more potent or selective antifungal drugs or even medicines lowering cholesterol in humans.
Authors: M Nowosielski, M Hoffmann, LS Wyrwicz, P Stepniak, D Plewczynski, M Lazniewski, K Ginalski, L Rychlewski
Note: 'Detailed mechanism of squalene epoxidase inhibition by terbinafine' by M. Nowosielski, M. Hoffmann, LS Wyrwicz, P. Stepniak, D. Plewczynski, M. Lazniewski, K. Ginalski, L. Rychlewski. J Chem Inf Model 51(2):455-62 (2011).
2010
Abstract: The 'Brainstorming' approach presented in this paper is a weighted voting method that can improve the quality of predictions generated by several machine learning (ML) methods. First, an ensemble of heterogeneous ML algorithms is trained on available experimental data, then all solutions are gathered and a consensus is built between them. The final prediction is performed using a voting procedure, whereby the vote of each method is weighted according to a quality coefficient calculated using multivariable linear regression (MLR). The MLR optimization procedure is very fast, therefore no additional computational cost is introduced by using this jury approach. Here, brainstorming is applied to selecting actives from large collections of compounds relating to five diverse biological targets of medicinal interest, namely HIV-reverse transcriptase, cyclooxygenase-2, dihydrofolate reductase, estrogen receptor, and thrombin. The MDL Drug Data Report (MDDR) database was used for selecting known inhibitors for these protein targets, and experimental data was then used to train a set of machine learning methods. The benchmark dataset (available at http://bio.icm.edu.pl/∼darman/chemoinfo/benchmark.tar.gz ) can be used for further testing of various clustering and machine learning methods when predicting the biological activity of compounds. Depending on the protein target, the overall recall value is raised by at least 20% in comparison to any single machine learning method (including ensemble methods like random forest) and unweighted simple majority voting procedures.
Authors: D Plewczynski
Note: 'Brainstorming: weighted voting prediction of inhibitors for protein targets' by D. Plewczynski. Journal of Molecular Modelling, Epub, Sept 21 (2010);
Abstract: Steroid-related cancers can be treated by inhibitors of steroid metabolism. In searching for new inhibitors of human 17beta-hydroxysteroid dehydrogenase type 1 (17beta-HSD 1) for the treatment of breast cancer or endometriosis, novel substances based on 15-substituted estrone were validated. We checked the specificity for different 17beta-HSD types and species. Compounds were tested for specificity in vitro not only towards recombinant human 17beta-HSD types 1, 2, 4, 5 and 7 but also against 17beta-HSD 1 of several other species including marmoset, pig, mouse, and rat. The latter are used in the processes of pharmacophore screening. We present the quantification of inhibitor preferences between human and animal models. Profound differences in the susceptibility to inhibition of steroid conversion among all 17beta-HSDs analyzed were observed. Especially, the rodent 17beta-HSDs 1 were significantly less sensitive to inhibition compared to the human ortholog, while the most similar inhibition pattern to the human 17beta-HSD 1 was obtained with the marmoset enzyme. Molecular docking experiments predicted estrone as the most potent inhibitor. The best performing compound in enzymatic assays was also highly ranked by docking scoring for the human enzyme. However, species-specific prediction of inhibitor performance by molecular docking was not possible. We show that experiments with good candidate compounds would out-select them in the rodent model during preclinical optimization steps. Potentially active human-relevant drugs, therefore, would no longer be further de
Authors: G Moeller, B Husen, D Kowalik, L Hirvela, D Plewczynski, L Rychlewski, J Messinger, H Thole, and J Adamski
Note: 'Species-Dependent Susceptibility of Inhibition of 17beta-Hydroxysteroid Dehydrogenase Type 1: Enzyme Inhibition and Molecular Docking Study' by G. Moeller, B. Husen, D. Kowalik, L. Hirvela, D. Plewczynski, L. Rychlewski, Suppl. 1 JUN (2010)
Abstract: Background: We present here the recent update of AMS algorithm for identification of post-translational modification (PTM) sites in proteins based only on sequence information, using artificial neural network (ANN) method. The query protein sequence is dissected into overlapping short sequence segments. Ten different physicochemical features describe each amino acid; therefore nine residues long segment is represented as a point in a 90 dimensional space. The database of sequence segments with confirmed by experiments post-translational modification sites are used for training a set of ANNs. Results: The efficiency of the classification for each type of modification and the prediction power of the method is estimated here using recall (sensitivity), precision values, the area under receiver operating characteristic (ROC) curves and leave-one-out tests (LOOCV). The significant differences in the performance for differently optimized neural networks are observed, yet the AMS 3.0 tool integrates those heterogeneous classification schemes into the single consensus scheme, and it is able to boost the precision and recall values independent of a PTM type in comparison with the currently available state-of-the art methods. Conclusions: The standalone version of AMS 3.0 presents an efficient way to indentify post-translational modifications for whole proteomes. The training datasets, precompiled binaries for AMS 3.0 tool and the source code are available at http://code.google.com/p/automotifserver under the Apache 2.0 license scheme.
Authors: S Basu and D Plewczynski
Note: 'AMS 3.0: prediction of post-translational modifications' by S. Basu and D. Plewczynski. BMC Bioinformatics, Epub, Apr 28, 11:210, (2010);
Abstract: Molecular docking is a widely used method for lead optimization. However, docking tools often fail to predict how a ligand (the smaller molecule, such as a substrate or drug candidate) binds to a receptor (the accepting part of a protein). We present here the HarmonyDOCK, a novel method for assessing the docking software accuracy, and creating the scoring function which would determine consensus protein-ligand pose among those generated by available docking programs. Conformations for few hundred protein-ligand complexes with known three-dimensional structure were predicted on a benchmark set by set of different docking programs. On the basis of the derived ranking, the point of reference and the lower score limit were determined for subsequent investigations. The focus of the methodology is on the top-ranked poses, with the assumption being that the conformation of the docked molecules is the most accurate. We found out that some docking programs perform considerably better than the others, yet in all cases the proper selection of decoys, namely HarmonyDOCK, is needed for successful docking procedure.
Authors: D Plewczynski, A Philips, M von Grotthuss, M Łaźniewski, L Rychlewski and K Ginalski
Note: 'HarmonyDOCK: The structural analysis of poses in protein- ligand docking' by D. Plewczynski, A. Philips, M. von Grotthuss, M. Łaźniewski, L. Rychlewski and K. Ginalski. Journal of Computational Biology, Epub, Nov 20 (2010);
Abstract: Steroid-related cancers can be treated by inhibitors of steroid metabolism. In searching for new inhibitors of human 17beta-hydroxysteroid dehydrogenase type 1 (17beta-HSD 1) for the treatment of breast cancer or endometriosis, novel substances based on 15-substituted estrone were validated. We checked the specificity for different 17beta-HSD types and species. Compounds were tested for specificity in vitro not only towards recombinant human 17beta-HSD types 1, 2, 4, 5 and 7 but also against 17beta-HSD 1 of several other species including marmoset, pig, mouse, and rat. The latter are used in the processes of pharmacophore screening. We present the quantification of inhibitor preferences between human and animal models. Profound differences in the susceptibility to inhibition of steroid conversion among all 17beta-HSDs analyzed were observed. Especially, the rodent 17beta-HSDs 1 were significantly less sensitive to inhibition compared to the human ortholog, while the most similar inhibition pattern to the human 17beta-HSD 1 was obtained with the marmoset enzyme. Molecular docking experiments predicted estrone as the most potent inhibitor. The best performing compound in enzymatic assays was also highly ranked by docking scoring for the human enzyme. However, species-specific prediction of inhibitor performance by molecular docking was not possible. We show that experiments with good candidate compounds would out-select them in the rodent model during preclinical optimization steps. Potentially active human-relevant drugs, therefore, would no longer be further developed. Activity and efficacy screens in heterologous species systems must be evaluated with caution.
Authors: G Möller, B Husen, D Kowalik, L Hirvelä, D Plewczyński, L Rychlewski, J Messinger, H Thole, J Adamski
Note: 'Species used for drug testing reveal different inhibition susceptibility for 17beta- hydroxysteroid dehydrogenase type 1' by G. Möller, B. Husen, D. Kowalik, L. Hirvelä, D. Plewczyński, L. Rychlewski, J. Messinger, H. Thole, J. Adamski, PLoS ONE Jun 8;5(6):e10969 (2010);
Authors: I Saha, U Maulik, S Bandyopadhyay and D. Plewczynski
Note: 'SVM ensemble crisp clustering for IRS image segmentation' by I. Saha, U. Maulik, S. Bandyopadhyay and D. Plewczynski. IEEEGRS LETTER (2010);
2009
Abstract: Computational screening of compound databases has become increasingly popular in pharmaceutical research. This review focuses on the evaluation of ligand-based virtual screening using active compounds as templates in the context of drug discovery. Ligand-based screening techniques are based on comparative molecular similarity analysis of compounds with known and unknown activity. We provide an overview of publications that have evaluated different machine learning methods, such as support vector machines, decision trees, ensemble methods such as boosting, bagging and random forests, clustering methods, neuronal networks, naïve Bayesian, data fusion methods and others.
Authors: D Plewczynski, S Spieser and U Koch
Note: 'Performance of machine learning methods for ligand-based virtual screening' by D. Plewczynski, S. Spieser and U. Koch. Combinatorial Chemistry & High Throughput Screening CCHTS 12(4): 358-68 (2009);
Abstract: We discuss here the mean-field theory for a cellular automata model of meta-learning. The meta-learning is the process of combining outcomes of individual learning procedures in order to determine the final decision with higher accuracy than any single learning method. Our method is constructed from an ensemble of interacting, learning agents, that acquire and process incoming information using various types, or different versions of machine learning algorithms. The abstract learning space, where all agents are located, is constructed here using a fully connected model that couples all agents with random strength values. The cellular automata network simulates the higher level integration of information acquired from the independent learning trials. The final classification of incoming input data is therefore defined as the stationary state of the meta-learning system using simple majority rule, yet the minority clusters that share opposite classification outcome can be observed in the system. Therefore, the probability of selecting proper class for a given input data, can be estimated even without the prior knowledge of its affiliation. The fuzzy logic can be easily introduced into the system, even if learning agents are build from simple binary classification machine learning algorithms by calculating the percentage of agreeing agents.
Authors: D Plewczynski
Note: 'Mean-field theory of meta-learning' by D. Plewczynski. J. Stat. Mech. P11003 (2009)
Abstract: Protein targets specificity classification is an important step in computational drug development and design efforts. The enhanced classification models of small chemical molecules enable the rapid scanning of large compounds databases. Here, we present the k-nearest neighbors with genetic algorithm feature optimization approach for selection of small molecule protein inhibitors. The method is trained on selected, diverse activity classes of the MDL drug data report (MDDR) with ligands described using simple atom pairs two dimensional chemical descriptors. The accuracy of inhibitors identification is presented in confusion tables with calculated recall and precision values. The precision for selected types of targets exceeded 70%, and the recall reaches 40%. As a consequence, the method can be easily applied to large commercial compounds collections in a drug development campaign in order to significantly reduce the number of ligands for further costly experimental validation.
Authors: D Plewczynski
Note: 'kNNsim: k-Nearest neighbors similarity with genetic algorithm features optimization enhances the prediction of activity classes for small molecules' by D. Plewczynski. Journal of Molecular Modelling 15, 591-596 (2009);
Abstract: We present here the estimation of the upper limit of the number of molecular targets in the human genome that represent an opportunity for further therapeutic treatment. We select around approximately 6300 human proteins that are similar to sequences of known protein targets collected from DrugBank database. Our bioinformatics study estimates the size of 'druggable' human genome to be around 20% of human proteome, i.e. the number of the possible protein targets for small-molecule drug design in medicinal chemistry. We do not take into account any toxicity prediction, the three-dimensional characteristics of the active site in the predicted 'druggable' protein families, or detailed chemical analysis of known inhibitors/drugs. Instead we rely on remote homology detection method Meta-BASIC, which is based on sequence and structural similarity. The prepared dataset of all predicted protein targets from human genome presents the unique opportunity for developing and benchmarking various in silico chemo/bio-informatics methods in the context of the virtual high throughput screening.
Authors: D Plewczynski and L Rychlewski
Note: 'Meta-basic estimates the size of druggable human genome' by D. Plewczynski and L. Rychlewski. Journal of Molecular Modelling 15, 695-699 (2009);
Abstract: We present here the random forest supervised machine learning algorithm applied to flexible docking results from five typical virtual high throughput screening (HTS) studies. Our approach is aimed at: i) reducing the number of compounds to be tested experimentally against the given protein target and ii) extending results of flexible docking experiments performed only on a subset of a chemical library in order to select promising inhibitors from the whole dataset. The random forest (RF) method is applied and tested here on compounds from the MDL drug data report (MDDR). The recall values for selected five diverse protein targets are over 90% and the performance reaches 100%. This machine learning method combined with flexible docking is capable to find 60% of the active compounds for most protein targets by docking only 10% of screened ligands. Therefore our in silico approach is able to scan very large databases rapidly in order to predict biological activity of small molecule inhibitors and provides an effective alternative for more computationally demanding methods in virtual HTS.
Authors: D Plewczynski, Marcin von Grotthuss, L Rychlewski, and Krzysztof Ginalski
Note: 'Virtual High-Throughput Screening using combined Random Forest and Flexible Docking' by D. Plewczynski, Marcin von Grotthuss, L. Rychlewski, and Krzysztof Ginalski. Combinatorial Chemistry & High Throughput Screening, CCHTS 12(5), p:484-9 (2009);
Abstract: The term Interactome describes the set of all molecular interactions in cells, especially in the context of protein-protein interactions. These interactions are crucial for most cellular processes, so the full representation of the interaction repertoire is needed to understand the cell molecular machinery at the system biology level. In this short review, we compare various methods for predicting protein-protein interactions using sequence and structure information. The ultimate goal of those approaches is to present the complete methodology for the automatic selection of interaction partners using their amino acid sequences and/or three dimensional structures, if known. Apart from a description of each method, details of the software or web interface needed for high throughput prediction on the whole genome scale are also provided. The proposed validation of the theoretical methods using experimental data would be a better assessment of their accuracy.
Authors: D Plewczynski and K Ginalski
Note: 'INTERACTOME: the prediction of protein-protein interactions in a cell' by D. Plewczynski and K. Ginalski. Cellular & Molecular Biology Letters Cell Mol Biol Lett. 14(1):1-22 (2009);
2008
Authors: D Plewczynski, A Tkacz, K Ginalski and L Rychlewski
Note: '2007 update of AutoMotif Server with reference database for prediction of Post-Translational Modification sites in proteins by Various Machine Learning Methods' by D. Plewczynski, A. Tkacz, K. Ginalski and L. Rychlewski. Journal of Molecular Modelling 14 (1), p:69-76 (2008);
Abstract: The ‘omics’ revolution is causing a flurry of data that all needs to be annotated for it to become useful. Sequences of proteins of unknown function can be annotated with a putative function by comparing them with proteins of known function. This form of annotation is typically performed with BLAST or similar software. Structural genomics is nowadays also bringing us three dimensional structures of proteins with unknown function. We present here software that can be used when sequence comparisons fail to determine the function of a protein with known structure but unknown function. The software, called 3D-Fun, is implemented as a server that runs at several European institutes and is freely available for everybody at all these sites. The 3D-Fun servers accept protein coordinates in the standard PDB format and compare them with all known protein structures by 3D structural superposition using the 3D-Hit software. If structural hits are found with proteins with known function, these are listed together with their function and some vital comparison statistics. This is conceptually very similar in 3D to what BLAST does in 1D. Additionally, the superposition results are displayed using interactive graphics facilities. Currently, the 3D-Fun system only predicts enzyme function but an expanded version with Gene Ontology predictions will be available soon. The server can be accessed at http://3dfun.bioinfo.pl/ or at http://3dfun.cmbi.ru.nl/.
Authors: M von Grotthuss, D Plewczynski, G Vriend, and L Rychlewski
Note: '3D-Fun: predicting enzyme function from structure' by M. von Grotthuss, D. Plewczynski, G. Vriend, and L. Rychlewski. Nucleic Acids Res. 36(Web Server issue), W303-7 (2008);
Abstract: We present here a neural network-based method for detection of signal peptides (abbreviation used: SP) in proteins. The method is trained on sequences of known signal peptides extracted from the Swiss-Prot protein database and is able to work separately on prokaryotic and eukaryotic proteins. A query protein is dissected into overlapping short sequence fragments, and then each fragment is analyzed with respect to the probability of it being a signal peptide and containing a cleavage site. While the accuracy of the method is comparable to that of other existing prediction tools, it provides a significantly higher speed and portability. The accuracy of cleavage site prediction reaches 73% on heterogeneous source data that contains both prokaryotic and eukaryotic sequences while the accuracy of discrimination between signal peptides and non-signal peptides is above 93% for any source dataset. As a consequence, the method can be easily applied to genome-wide datasets. The software can be downloaded freely from http://rpsp.bioinfo.pl/RPSP.tar.gz.
Authors: Dariusz Plewczynski, Lukasz Slabinski, Krzysztof Ginalski and Leszek Rychewski
Note: 'Prediction of signal peptides in protein sequences by neural networks' by Dariusz Plewczynski, Lukasz Slabinski, Krzysztof Ginalski and Leszek Rychewski. Acta Biochimica Polonica 55(2), p. 261-7 (2008);
2007
Abstract: A structure-based in silico virtual drug discovery procedure was assessed with severe acute respiratory syndrome coronavirus main protease serving as a case study. First, potential compounds were extracted from protein-ligand complexes selected from Protein Data Bank database based on structural similarity to the target protein. Later, the set of compounds was ranked by docking scores using a Electronic High-Throughput Screening flexible docking procedure to select the most promising molecules. The set of best performing compounds was then used for similarity search over the 1 million entries in the Ligand.Info Meta-Database. Selected molecules having close structural relationship to a 2-methyl-2,4-pentanediol may provide candidate lead compounds toward the development of novel allosteric severe acute respiratory syndrome protease inhibitors.
Authors: D Plewczynski, M Hoffmann, M von Grotthuss, K Ginalski, L Rychewski
Note: 'In Silico Prediction of SARS Protease Inhibitors' by D. Plewczynski, M. Hoffmann , M. von Grotthuss, K. Ginalski, L. Rychewski. Chemical Biology & Drug Design CBDD, 69 (4), p. 269 (2007);
Authors: L Knizewski, K, Steczkiewicz, K Kuchta, L Wyrwicz, D Plewczynski, A Kolinski, L Rychlewski, K Ginalski
Note: 'Uncharacterized DUF1574 leptospira proteins are SGNH hydrolases' by L. Knizewski, K, Steczkiewicz, K. Kuchta, L. Wyrwicz, D. Plewczynski, A. Kolinski, L. Rychlewski, K. Ginalski. Cell Cycle 7(4), p.542-4 (2007);
Abstract: The rapid increase in genomic information requires new automatic techniques to investigate protein functions. The function of proteins is partially determined by short sequence segments. For example the phosphorylation by protein kinases is an important mechanism for controlling intracellular processes. Many kinases are known, but the identification of their potential biological targets is still ongoing research. High substrate specificity of protein kinases ensures correct transmission of signals in cells. The specificity is largely determined by the primary sequence of the target site, but we lack general, efficient and error prune tools for identifying these sites. Most methods designed to predict functional motifs process local sequence information around post-translational modification sites. We present here an advanced computational protocol for rapid identification of post-translational modifications (PTM) in proteins on the whole genome scale. The AutoMotif Server (AMS) identifies various types of post-translational modifications in protein sequences. A query protein sequence is dissected into overlapping short segments. Each segment is projected into an abstract space of sequence fragments by 10 different representations. Those projections are compared with the database of representations of known and confirmed by experiments post-translational modification sites using the support vector machine (SVM) approach 1, 2. The supervised machine learning approach is able to predict the most of post-translational modification sites in proteins. It is based on the classification of the biological functional information acquired from the Swiss-Prot database version 4.2. The classification models are then used to predict new modification sites in proteins. Users can access a list of sites in proteins annotated as being able to undergo certain post-translational modification in Swiss-Prot database and add new annotated sequence segments from proteins (positive instances). The AMS server was demonstrated 3, 4 to gain high accuracy in distinguishing short sequence fragments that are post-translational modified from those that are not. The efficiency of the classification for each type of modifications and the prediction power of several versions of the method is estimated using the standardized leave-one-out tests. The sensitivities of the protocol for all types of modifications are in the range of 70%. The AutoMotif Server is freely available at http://automotif.bioinfo.pl/. The local version of the software is available on request from the authors. The parameters (the search type, the number of top models, and the PTM type) are optional and can be easily modified. The following protocol describes how to use AMS server to detect various types of post-translational modifications, and how to understand the resulting score for a given prediction.
Authors: D Plewczynski A Tkacz
Note: 'AutoMotif Server: A Computational Protocol for Identification of Post-Translational Modifications in Protein Sequences' D. Plewczynski A. Tkacz. Nature Protocols doi: 10.1038/ nprot.2007.183 (2007);
Abstract: The annotation of protein folds within newly sequenced genomes is the main target for semi-automated protein structure prediction (virtual structural genomics). A large number of automated methods have been developed recently with very good results in the case of single-domain proteins. Unfortunately, most of these automated methods often fail to properly predict the distant homology between a given multi-domain protein query and structural templates. Therefore a multi-domain protein should be split into domains in order to overcome this limitation. ProteinSplit is designed to identify protein domain boundaries using a novel algorithm that predicts disordered regions in protein sequences. The software utilizes various sequence characteristics to assess the local propensity of a protein to be disordered or ordered in terms of local structure stability. These disordered parts of a protein are likely to create interdomain spacers. Because of its speed and portability, the method was successfully applied to several genome-wide fold annotation experiments. The user can run an automated analysis of sets of proteins or perform semi-automated multiple user projects (saving the results on the server). Additionally the sequences of predicted domains can be sent to the Bioinfo.PL Protein Structure Prediction Meta-Server for further protein three-dimensional structure and function prediction. The program is freely accessible as a web service at http://lucjan.bioinfo.pl/proteinsplit together with detailed benchmark results on the critical assessment of a fully automated structure prediction (CAFASP) set of sequences. The source code of the local version of protein domain boundary prediction is available upon request from the authors.
Authors: Lucjan Wyrwicz, Grzegorz Koczyk, Leszek Rychlewski and Dariusz Plewczynski
Note: 'ProteinSplit: splitting of multi-domain using prediction of ordered and disordered regions of protein sequences for virtual structural genomics' by Lucjan Wyrwicz, Grzegorz Koczyk, Leszek Rychlewski and Dariusz Plewczynski. Journal of Physics: Condensed Matter, Special Issue: Structure and Function of Biomolecules 19, p. 285222 (2007);
Abstract: In many cases at the beginning of an HTS-campaign, some information about active molecules is already available. Often known active compounds (such as substrate analogues, natural products, inhibitors of a related protein or ligands published by a pharmaceutical company) are identified in low-throughput validation studies of the biochemical target. In this study we evaluate the effectiveness of a support vector machine applied for those compounds and used to classify a collection with unknown activity. This approach was aimed at reducing the number of compounds to be tested against the given target. Our method predicts the biological activity of chemical compounds based on only the atom pairs (AP) two dimensional topological descriptors. The supervised support vector machine (SVM) method herein is trained on compounds from the MDL drug data report (MDDR) known to be active for specific protein target. For detailed analysis, five different biological targets were selected including cyclooxygenase-2, dihydrofolate reductase, thrombin, HIV-reverse transcriptase and antagonists of the estrogen receptor. The accuracy of compound identification was estimated using the recall and precision values. The sensitivities for all protein targets exceeded 80% and the classification performance reached 100% for selected targets. In another application of the method, we addressed the absence of an initial set of active compounds for a selected protein target at the beginning of an HTS-campaign. In such a case, virtual high-throughput screening (vHTS) is usually applied by using a flexible docking procedure. However, the vHTS experiment typically contains a large percentage of false positives that should be verified by costly and time-consuming experimental follow-up assays. The subsequent use of our machine learning method was found to improve the speed (since the docking procedure was not required for all compounds from the database) and also the accuracy of the HTS hit lists (the enrichment factor).
Authors: D Plewczynski, L Rychlewski, Marcin von Grotthuss, Stephane Spieser, Leszek Rychewski, Lucjan S Wyrwicz, Krzysztof Ginalski and Uwe Koch
Note: 'Target Specific Compound Identification using Support Vector Machine.' by D. Plewczynski, L. Rychlewski, Marcin von Grotthuss, Stephane Spieser, Leszek Rychewski, Lucjan S. Wyrwicz, Krzysztof Ginalski and Uwe Koch. Combinatorial Chemistry & High Throughput Screening CCHTS, 10(3), p. 189-196(8) (2007);
Abstract: DNA microarrays are the modern, parallel version of classic molecular biology hybridization techniques allowing the exploration of thousands of sequences in a single run. The technique permits the complete analysis of genetic material and the monitoring of expression changes occurring in a biological sample under various conditions. Microarrays have been used successfully in various research areas including: sequencing, single nucleotide polymorphism (SNP) detection, characterization of protein-DNA interactions, DNA computing, mRNA profiling, and many more. Applications of microarrays in the biosciences include: gene expression studies, disease diagnosis, pharmacogenomics, drug screening, pathogen detection, and genotyping. This book focuses on the current successful applications of microarrays in various areas of bioscience. The authors describe the use of microarrays to assist basic research and drug discovery by the generation of gene expression maps, the reconstruction of gene networks and the study of biochemical pathways, the classification of genes and biological samples, and a real case study on the transcriptional effects of vaccine treatment. An entire chapter is dedicated to applications of microarrays that do not involve gene expression. This book is designed for researchers and students in all areas of biosciences.
Authors: Adrian Tkacz, Leszek Rychlewski, Paolo Uva, Dariusz Plewczyński
Note: 'Supervised Classification of Genes and Biological Samples' by Adrian Tkacz, Leszek Rychlewski, Paolo Uva, Dariusz Plewczyński. Chapter in book 'DNA Microarrays. Current Applications', pages 101-119, Horizon Bioscience (2007);
Abstract: In many cases, at the beginning of a high throughput screening experiment some information about active molecules is already available. Active compounds (such as substrate analogues, natural products and inhibitors of related proteins) are often identified in low throughput validation studies on a biochemical target. Sometimes the additional structural information is also available from crystallographic studies on protein and ligand complexes. In addition, the structural or sequence similarity of various protein targets yields a novel possibility for drug discovery. Co-crystallized compounds from homologous proteins can be used to design leads for a new target without co-crystallized ligands. In this paper we evaluate how far such an approach can be used in a real drug campaign, with severe acute respiratory syndrome (SARS) coronavirus providing an example. Our method is able to construct small molecules as plausible inhibitors solely on the basis of the set of ligands from crystallized complexes of a protein target, and other proteins from its structurally homologous family. The accuracy and sensitivity of the method are estimated here by the subsequent use of an electronic high throughput screening flexible docking algorithm. The best performing ligands are then used for a very restrictive similarity search for potential inhibitors of the SARS protease within the million compounds from the Ligand.Info small molecule meta-database. The selected molecules can be passed on for further experimental validation.
Authors: Dariusz Plewczynski, Marcin Hoffmann, Marcin von Grotthuss, Lukasz Knizewski, Leszek Rychlewski, Krystian Eitner and Krzysztof Ginalski
Note: 'Modelling of potentially promising SARS protease inhibitors' by Dariusz Plewczynski, Marcin Hoffmann, Marcin von Grotthuss, Lukasz Knizewski, Leszek Rychlewski, Krystian Eitner and Krzysztof Ginalski. Journal of Physics: Condensed Matter, Special Issue: Structure and Function of Biomolecules 19, p. 285207 (2007);
Abstract: The RPSP is a fast web service for detection of signal peptides in proteins. The method uses neural networks trained on known signal peptides from the Swiss-Prot protein database. The web server works either on prokaryotic and eukaryotic proteins or without specifying an organism type. The accuracy of the web server is similar to other available computational prediction web services, yet because of its speed and portability the method can be easily applied to whole proteomes. The RPSP web server is available at http://rpsp.bioinfo.pl.
Authors: Dariusz Plewczynski, Lukasz Slabinski, Adrian Tkacz, Laszlo Kajan, Liisa Holm, Krzysztof Ginalski and Leszek Rychewski
Note: 'The RPSP: Web server for prediction of signal peptides' by Dariusz Plewczynski, Lukasz Slabinski, Adrian Tkacz, Laszlo Kajan, Liisa Holm, Krzysztof Ginalski and Leszek Rychewski. Polymer 48, p. 5493-5496 (2007);
Authors: D Plewczynski
Note: 'How to estimate the size of Druggable Human Genome?' by D. Plewczynski. Advances in ChemInformatics 1, p. 11-19 (2007);
2006
Abstract: Our algorithm predicts short linear functional motifs in proteins using only sequence information. Statistical models for short linear functional motifs in proteins are built using the database of short sequence fragments taken from proteins in the current release of the Swiss-Prot database. Those segments are confirmed by experiments to have single-residue post-translational modification. The sensitivities of the classification for various types of short linear motifs are in the range of 70%. The query protein sequence is dissected into short overlapping fragments. All segments are represented as vectors. Each vector is then classified by a machine learning algorithm (Support Vector Machine) as potentially modifiable or not. The resulting list of plausible post-translational sites in the query protein is returned to the user. We also present a study of the human protein kinase C family as a biological application of our method.
Authors: D Plewczynski, A Tkacz, L Wyrwicz, A Godzik, A Kloczkowski and L Rychlewski
Note: 'Support Vector Machine Classification of Linear Functional Motifs in Proteins' by D. Plewczynski, A. Tkacz, L. Wyrwicz, A. Godzik, A. Kloczkowski and L. Rychlewski. Journal of Molecular Modelling 12(4), p. 453-61 (2006);
Abstract: Background: The number of protein structures from structural genomics centers dramatically increases in the Protein Data Bank (PDB). Many of these structures are functionally unannotated because they have no sequence similarity to proteins of known function. However, it is possible to successfully infer function using only structural similarity. Results: Here we present the PDB-UF database, a web-accessible collection of predictions of enzymatic properties using structure-function relationship. The assignments were conducted for three-dimensional protein structures of unknown function that come from structural genomics initiatives. We show that 4 hypothetical proteins (with PDB accession codes: 1VH0, 1NS5, 1O6D, and 1TO0), for which standard BLAST tools such as PSI-BLAST or RPS-BLAST failed to assign any function, are probably methyltransferase enzymes. Conclusion: We suggest that the structure-based prediction of an EC number should be conducted having the different similarity score cutoff for different protein folds. Moreover, performing the annotation using two different algorithms can reduce the rate of false positive assignments. We believe, that the presented web-based repository will help to decrease the number of protein structures that have functions marked as 'unknown' in the PDB file.
Authors: M von Grotthuss, D Plewczynski, K Ginalski, L Rychlewski, and EI Shakhnovich
Note: 'PDB UF: database of predicted enzymatic functions for unannotated protein structures from Structural Genomics' by M. von Grotthuss, D. Plewczynski, K. Ginalski, L. Rychlewski, and E.I. Shakhnovich. BMC Bioinformatics 6(7), p. 53 (2006);
Abstract: How well do different classification methods perform in selecting the ligands of a protein target out of large compound collections not used to train the model? Support vector machines, random forest, artificial neural networks, k-nearest-neighbor classification with genetic-algorithm-optimized feature selection, trend vectors, naïve Bayesian classification, and decision tree were used to divide databases into molecules predicted to be active and those predicted to be inactive. Training and predicted activities were treated as binary. The database was generated for the ligands of five different biological targets which have been the object of intense drug discovery efforts: HIV-reverse transcriptase, COX2, dihydrofolate reductase, estrogen receptor, and thrombin. We report significant differences in the performance of the methods independent of the biological target and compound class. Different methods can have different applications; some provide particularly high enrichment, others are strong in retrieving the maximum number of actives. We also show that these methods do surprisingly well in predicting recently published ligands of a target on the basis of initial leads and that a combination of the results of different methods in certain cases can improve results compared to the most consistent method.
Authors: D Plewczynski, S Spieser, U Koch
Note: 'Assessing Different Classification Methods for Virtual Screening' by D. Plewczynski, S. Spieser, U. Koch. J. Chem. Inf. Model. 46(3), p.1098-106 (2006);
Abstract: The Dali program is widely used for carrying out automatic comparisons of protein structures determined by X-ray crystallography or NMR. The most familiar version is the Dali server, which performs a database search comparing a query structure supplied by the user against the database of known structures (PDB) and returns the list of structural neighbors by e-mail. The more recently introduced DaliLite server compares two structures against each other and visualizes the result interactively. The Dali database is a structural classification based on precomputed all-against-all structural similarities within the PDB. The resulting hierarchical classification can be browsed on the Web and is linked to protein sequence classification resources. All Dali resources use an identical algorithm for structure comparison. Users may run Dali using the Web, or the program may be downloaded to be run locally on Linux computers.
Authors: L Holm, S Kääriäinen, C Wilton and D Plewczynski
Note: 56)'Using Dali for Structural Comparison of Proteins' by L. Holm, S. Kääriäinen, C. Wilton and D. Plewczynski. Current Protocols in Bioinformatics 5, p. 5.5.1-5.5.24 (2006),
2005
Abstract: A new bioinformatics tool for molecular modeling of the local structure around phosphorylation sites in proteins has been developed. Our method is based on a library of short sequence and structure motifs. The basic structural elements to be predicted are local structure segments (LSSs). This enables us to avoid the problem of non-exact local description of structures, caused by either diversity in the structural context, or uncertainties in prediction methods. We have developed a library of LSSs and a profile--profile-matching algorithm that predicts local structures of proteins from their sequence information. Our fragment library prediction method is publicly available on a server (FRAGlib), at http://ffas.ljcrf.edu/Servers/frag.html . The algorithm has been applied successfully to the characterization of local structure around phosphorylation sites in proteins. Our computational predictions of sequence and structure preferences around phosphorylated residues have been confirmed by phosphorylation experiments for PKA and PKC kinases. The quality of predictions has been evaluated with several independent statistical tests. We have observed a significant improvement in the accuracy of predictions by incorporating structural information into the description of the neighborhood of the phosphorylated site. Our results strongly suggest that sequence information ought to be supplemented with additional structural context information (predicted with our segment similarity method) for more successful predictions of phosphorylation sites in proteins.
Authors: D Plewczynski, L Jaroszewski, A Godzik, A Kloczkowski and L Rychlewski
Note: 'Molecular Modelling of Phosphorylation Sites in Proteins using Database of Local Structure Segments' by D. Plewczynski, L. Jaroszewski, A. Godzik, A. Kloczkowski and L. Rychlewski. Journal of Molecular Modelling 11(6), p. 431-438 (2005);
Abstract: The AutoMotif Server allows for identification of post-translational modification (PTM) sites in proteins based only on local sequence information. The local sequence preferences of short segments around PTM residues are described here as linear functional motifs (LFMs). Sequence models for all types of PTMs are trained by support vector machine on short-sequence fragments of proteins in the current release of Swiss-Prot database (phosphorylation by various protein kinases, sulfation, acetylation, methylation, amidation, etc.). The accuracy of the identification is estimated using the standard leave-one-out procedure. The sensitivities for all types of short LFMs are in the range of 70%.
Authors: D Plewczynski, A Tkacz, L Wyrwicz and L Rychlewski
Note: 'AutoMotif Server: prediction of single residue post-translational modifications in proteins' by D. Plewczynski, A. Tkacz, L. Wyrwicz and L. Rychlewski. Bioinformatics 21(10), p.2525-7 (2005);
Abstract: We describe a bioinformatics tool that can be used to predict the position of phosphorylation sites in proteins based only on sequence information. The method uses the support vector machine (SVM) statistical learning theory. The statistical models for phosphorylation by various types of kinases are built using a dataset of short (9-amino acid long) sequence fragments. The sequence segments are dissected around post-translationally modified sites of proteins that are on the current release of the Swiss-Prot database, and that were experimentally confirmed to be phosphorylated by any kinase. We represent them as vectors in a multidimensional abstract space of short sequence fragments. The prediction method is as follows. First, a given query protein sequence is dissected into overlapping short segments. All the fragments are then projected into the multidimensional space of sequence fragments via a collection of different representations. Those points are classified with pre-built statistical models (the SVM method with linear, polynomial and radial kernel functions) either as phosphorylated or inactive ones. The resulting list of plausible sites for phosphorylation by various types of kinases in the query protein is returned to the user. The efficiency of the method for each type of phosphorylation is estimated using leave-one-out tests and presented here. The sensitivities of the models can reach over 70%, depending on the type of kinase. The additional information from profile representations of short sequence fragments helps in gaining a higher degree of accuracy in some phosphorylation types. The further development of an automatic phosphorylation site annotation predictor based on our algorithm should yield a significant improvement when using statistical algorithms in order to quantify the results.
Authors: D Plewczynski, A Tkacz, A Godzik and L Rychlewski
Note: 'A support vector machine approach to the identification of phosphorylation sites' by D. Plewczynski, A. Tkacz, A. Godzik and L. Rychlewski. Cellular & Molecular Biology Letters 10 (1): 73-89 (2005);
2004
Authors: D Plewczynski, J Pas, M von Grotthuss and L Rychlewski
Note: 'The Tool for Comparison of Proteins Based on the Segment Structural Similarity' by D. Plewczynski, J. Pas, M. von Grotthuss and L. Rychlewski. Acta Biochimica Polonica 51(1), p. 161-172 (2004);
Abstract: Backgroud: Defining blocks forming the global protein structure on the basis of local structural regularity is a very fruitful idea, extensively used in description, and prediction of structure from only sequence information. Over many years the secondary structure elements were used as available building blocks with great success. Specially prepared sets of possible structural motifs can be used to describe similarity between very distant, non-homologous proteins. The reason for utilizing the structural information in the description of proteins is straightforward. Structural comparison is able to detect approximately twice as many distant relationships as sequence comparison at the same error rate. Results: Here we provide a new fragment library for Local Structure Segment (LSS) prediction called FRAGlib which is integrated with a previously described segment alignment algorithm SEA. A joined FRAGlib/SEA server provides easy access to both algorithms, allowing a one stop alignment service using a novel approach to protein sequence alignment based on a network matching approach. The FRAGlib used as secondary structure prediction achieves only 73% accuracy in Q3 measure, but when combined with the SEA alignment, it achieves a significant improvement in pairwise sequence alignment quality, as compared to previous SEA implementation and other public alignment algorithms. The FRAGlib algorithm takes ~2 min. to search over FRAGlib database for a typical query protein with 500 residues. The SEA service align two typical proteins within circa ~5 min. All supplementary materials (detailed results of all the benchmarks, the list of test proteins and the whole fragments library) are available for download on-line at http://ffas.ljcrf.edu/darman/results/. Conclusions: The joined FRAGlib/SEA server will be a valuable tool both for molecular biologists working on protein sequence analysis and for bioinformaticians developing computational methods of structure prediction and alignment of proteins.
Authors: D Plewczynski, L Rychlewski, L Jaroszewski, Y Ye, A Godzik
Note: 'SEA and FRAGlib – an Integrated Web Service for Improving the Alignment Quality based on Segments Comparison' by D. Plewczynski, L. Rychlewski, L. Jaroszewski, Y. Ye, A. Godzik. BMC Bioinformatics 5(1):98 (2004);
2003
Abstract: We describe an ab initio server prototype for prediction of phosphorylation sites. A list of possible active sites for a given query protein is build using query protein sequence and the database of proteins annotated for a certain type of activation process by Swiss-Prot DB. All short segments of a query protein sequence centered around plausible active sites are compared with experimental profiles. Those profiles describe both sequence and structure preferences for each type of active site. Prediction of local conformation of a query protein chain around examined site is done with the specially prepared library of short local structural segments (LSSs). The short sequence fragments from a query protein are matched with segments in the library using profile with profile alignment. Predicted local structure of a chain near active site qualitatively agrees with experimental data fetched from PDB database. We estimate in this paper the level of improvement over purely sequence based methods gained by incorporating predicted structural information into the local description of phosphorylation sites.
Authors: D Plewczynski, L Rychlewski
Note: 'Ab Initio Server Prototype for Prediction of Phosphorylation Sites in Proteins' by D. Plewczynski, L. Rychlewski. Computational Methods in Science and Technology CMST vol 9(1-2), p. 93-100 (2003);
2002
Abstract: 3D-Hit is a fast scanning method for detecting structural similarities between proteins. The algorithm is based on a hashing function, which decomposes proteins into segments of 13 residues. The scanning procedures start with assigning a set of similar segments from the database to each segment in the query protein. These initial hits are expanded by two iterations of structural superposition of larger segments of 99 and 299 residues. The method generates an alignment for the query protein by concatenating partial structural alignments.
Authors: D Plewczynski, J Pas, M von Grotthuss, L Rychlewski
Note: '3D-Hit, Fast Structural Comparison of Proteins' by D. Plewczynski, J. Pas, M. von Grotthuss, L. Rychlewski. Applied Bioinformatics 1, p. 223-225 (2002);