STRUCTURAL GENOMICS
The nucleus of a cell contains chromatin - a complex of DNA and proteins that encodes the genes a living organism uses to carry out life. We are involved in the 4DNucleome project funded by the NIH, which is focused on understanding the higher order organisation of a nucleus and its functional consequences.
In the Laboratory of Functional and Structural Genomics we perform theoretical studies, whose main objective is to analyze and predict the three-dimensional structure of the human genome, and its relation with the genomic diversity of human populations, both natural and pathological. In particular, we investigate structural variants, copy number variants observed in various sub-populations and the groups of patients, and their three-dimensional localization in the structure of the nucleus.
Chromatin conformation capture experiments (ChIA-PET and Hi-C) give us information about loops and domains within the chromatin structure. On the other hand, experiments like ChIP-seq, GRO-seq, Bru-seq, ATAC-seq provide information about chromatin marks and DNA accessibility. Moreover microscopic data shows us the shape and the volume of the chromosomes and the DNA density inside their territory. We combine different type of the data and introduce them into the modelling for a better understanding of how chromatin structure determines function.
Multidimensional Monte Carlo
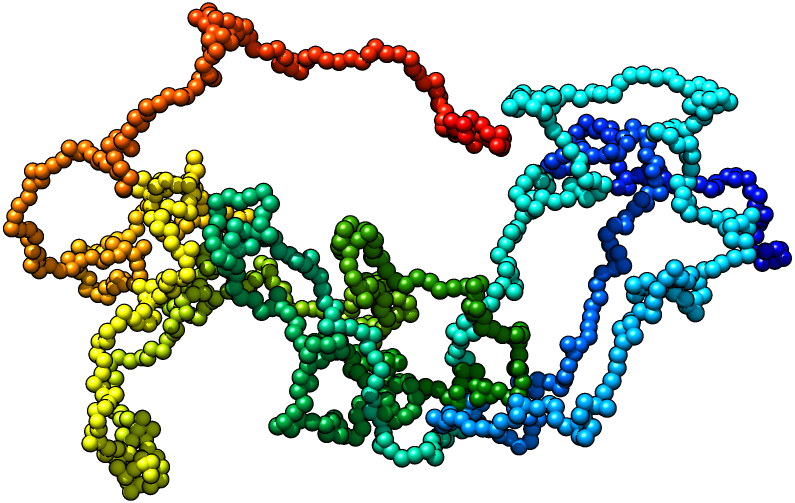
We developed a software tool called 3-Dimensional GeNOme Modeling Engine (3D-GNOME) to reconstruct the spatial chromatin conformation based on ChIA-PET data. We base our modeling on the underlying biological structures: chromatin loops and topological domains. First, we employ the weak interactions to create the low-resolution contact maps that we use to position topological domains in relation to each other. Then we take the advantage of the ChIA-PET specificity that allows to target a particular protein in order to identify a set of strong interactions indicating chromatin loops. In our modelling we also consider CTCF motifs orientation and weak interactions between individual chromatin loops. Taken together, this allows us to create reliable models of selected genomic regions, whole chromosomes and even whole genome in a reasonable time.
Multidimensional Scaling
We develop a method which uses a distance geometry tool — Multidimensional scaling (MDS) to reconstruct spatial structures of chromosomes from the distances between their elements. The approach consists of three major steps. Firstly, an experimentally driven interaction matrix is transformed into frequency-based, de-noised 2D map. Then, fuzzy graph distance map is calculated, with the use of a chosen algebra (i.e. T-norms), yielding another 2D map, which approximates Euclidean bead distances fairly. Eventually, MDS algorithm drives the data representation towards the three-dimensional structure of a chromosome (an ensemble), which is recovered. The latter is achieved by minimizing a cost function involving the deviations between the input distances and coordinates being reconstructed on the course of MDS simulation.
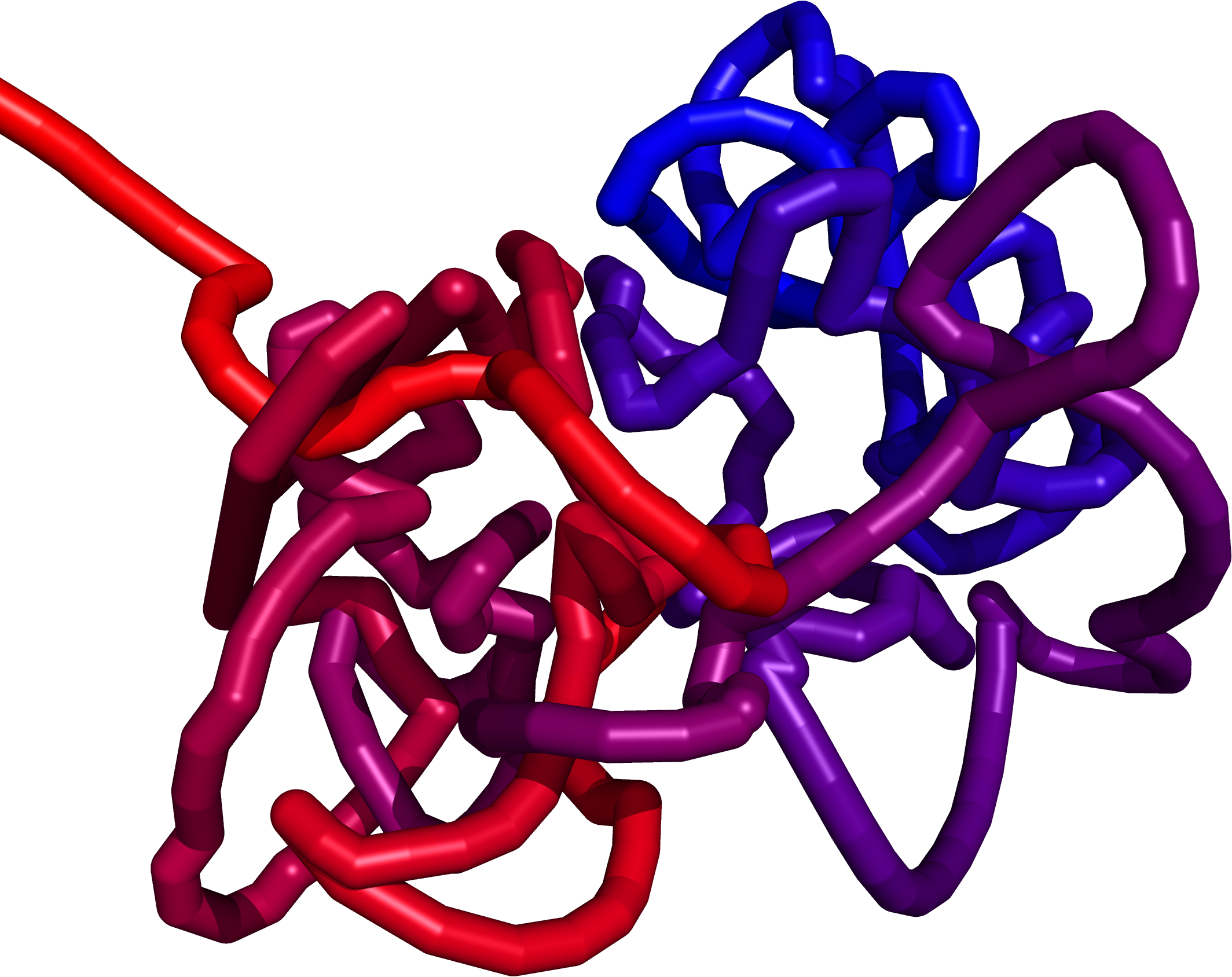
Multiscale Molecular Dynamics
The structure obtained from the Monte Carlo method creates a preliminary structure for the Multiscale Molecular Dynamics (MMD). Our new force field allow us to explore the chromatin with methods of molecular dynamics . Starting with a random polymer force field, we add additional parts to the potential energy function:
- Contacts from Hi-C and Chia-PET maps.
- Direct imaging methods obtained from confocal and electron microscopy.
- Genome compartmentalization from chromatin marks.
- DNA accessibility.
We believe that this approach will allow us to construct of a model fully compatible with the available experimental data so far. The force field is implemented in the GROMACS which provides high-scale parallelization with GPU support.
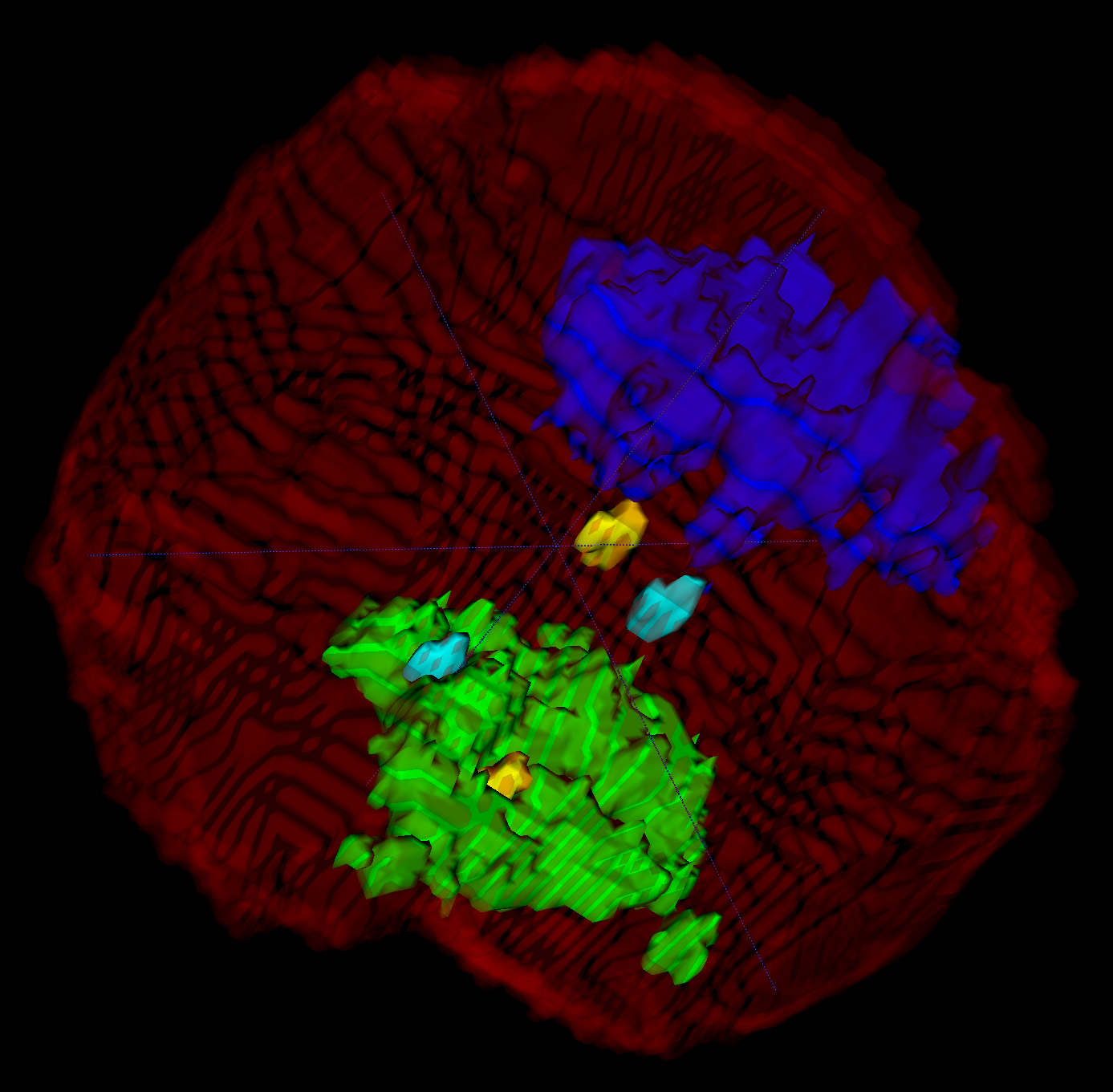
Confocal Microscopy
We work with data from confocal microscopy (high resolution optical microscopy). This data contain markers for chromatin density, chromosome 1 territory and telomere positions. We developed an algorithm to chose nuclear region from the raw image and second to do inner segmentation. Currently, we reconstruct the positions, shape and volume of two copies (paternal and maternal) of chromosome 1.
MEMBERS

Przemysław Szałaj
researcher
PhD candidate
Przemek Szałaj is interested in the functional and structural genomics, with a particular interest in the 3D chromatin organization. He developed simulation software for inferring the multiscale chromatin models using Hi-C and ChIA-PET data. Currently he works on applying the bioimaging data for refinement of the simulations. He is also interested in the variation of the structural genomic properties and their inheritance, with the ultimate goal being to better understand the significance of the genome folding on its function and the mechanisms that govern it.
E-mail: p.szalaj(at)cent.uw.edu.pl
Research areas: STRUCTURAL GENOMICS

Michał Sadowski
researcher
BSc
Michał Sadowski interests are from the field of physics and genome biology and are presently focused on genome architecture, genome organization and their link to gene regulation. These areas of study can be explored by an analysis of data coming from experimental methods which capture chromatin conformation both locally (3C, 4C) and genome-wide (Hi-C and ChIA-PET). This analysis can lead us to better understanding of gene expression mechanisms and provide a new insight into relation between gene expression and genomic variation. In his present work Michał Sadowski is trying to combine conformation capture data, genomic variation data and gene transcription data, in order to pursue more complete explanation of gene regulation processes, genome spatial arrangement and its evolution.
E-mail: m.sadowski(at)cent.uw.edu.pl
Research areas: STRUCTURAL GENOMICS

Grzegorz Bokota
researcher
MSc
Grzegorz Bokota interests are focused on developing algorithm for microscope image analyzing and massive parallel modeling of cell colony. In image analysis created algorithm are focused on nuclei segmentation and nuclei analysis of the interior of the nucleus.
E-mail: g.bokota(at)cent.uw.edu.pl
Research areas: STRUCTURAL GENOMICS , BIOLOGICAL SYSTEMS MODELING

Teresa Szczepińska
researcher
PhD
Teresa Szczepińska, PhD, is a bioinformatician. She has bachelor’s degree in molecular biology from Interfaculty Individual Studies in Mathematical and Natural Sciences at Warsaw University and master’s degree in bioinformatics from VU University Amsterdam. She accomplished doctoral degree at Nencki Institute of Experimental Biology, Polish Academy of Sciences. Teresa’s research interest is in genomic data, higher order chromatin organisation and its relation to transcription regulation. She is experienced in high throughput sequencing data analysis and in retrieving of biological information with it’s knowledge-based selection.
E-mail: t.szczepinska(at)cent.uw.edu.pl
Research areas: STRUCTURAL GENOMICS

Wayne Dawson
researcher
PhD
Wayne Dawson, PhD, is a physicist and structural biologist. He has been a postdoc in the Laboratory of Functional and Structural Genomics since April 2016. He received his PhD from The University of Tokyo. His main focus over the years has been on RNA/protein structure prediction and folding, electron transfer proteins, and currently, chromatin structure and problems related to the influenza virus.
E-mail: dawsonzhu(at)gmail.com
Research areas: STRUCTURAL GENOMICS

Michal Pietal
researcher
Executive DBA, PhD
Michal was researching transformations of chromatin 2D interaction matrices (Hi-C / ChIA-PET) into 3D models or ensembles, with the use of various 2D processing techniques, including fuzzy graph distance maps and Multi-dimensional Scaling (MDS). He was also involved into Hemagglutinin molecule de novo modelling, with the use of fuzzy contact maps of homologous templates from PDB. In addition, he was engaged in protein-protein interface classification from sequence data, with 2D maps prediction stage.
Research areas: STRUCTURAL GENOMICS

Ziad Al Bkhetan
researcher
PhD student
Ziad Al Bkhetan is a PhD student at University of Warsaw, He obtained his master degree in computer science from Warsaw University of Technology, Faculty of Mathematics and Information Science. He has good experience in Software Engineering, Data Mining and GPU Programming. Currently he is working on Genomic Interactions Prediction.
E-mail: z.albkhetan(at)cent.uw.edu.pl
Research areas: STRUCTURAL GENOMICS , BIOSTATISTICS AND COGNITIVE COMPUTING

Michał Kadlof
researcher
PhD student
Michał Kadlof graduated with a degree in bioinformatics and engineering studies in computer science with a specialization in database engineering. Currently he is doing a PhD in the Faculty of Physics at the University of Warsaw. His main interests are in computer simulations of the dynamics of biopolymers - proteins and nucleic acids. In the past, he dealt with protein structure prediction, and topology of proteins. Currently is conducting research on multiscale force fields that describe the behavior of chromatin within the nucleus. In addition, he is a computer systems administrator. He spends his free time doing electronics, hiking and medieval historical reenactments.
Tel: (+48 22) 55 43 752
E-mail: m.kadlof(at)cent.uw.edu.pl
Research areas: STRUCTURAL GENOMICS

Agnieszka Kraft
researcher
MSc student
Agnieszka Kraft is a student at the Bioinformatics and Systems Biology master's programme. Her interests are in genome architecture, especially structural variants and their link to phenotypes.
Research areas: STRUCTURAL GENOMICS
PAPERS
2016
Abstract: Recent advances in high-throughput chromosome conformation capture (3C) technology, such as Hi-C and ChIA-PET, have demonstrated the importance of 3D genome organization in development, cell differentiation and transcriptional regulation. There is now a widespread need for computational tools to generate and analyze 3D structural models from 3C data. Here we introduce our 3D GeNOme Modeling Engine (3D-GNOME), a web service which generates 3D structures from 3C data and provides tools to visually inspect and annotate the resulting structures, in addition to a variety of statistical plots and heatmaps which characterize the selected genomic region. Users submit a bedpe (paired-end BED format) file containing the locations and strengths of long range contact points, and 3D-GNOME simulates the structure and provides a convenient user interface for further analysis. Alternatively, a user may generate structures using published ChIA-PET data for the GM12878 cell line by simply specifying a genomic region of interest. 3D-GNOME is freely available at http://3dgnome.cent.uw.edu.pl/.
Authors: SzalajP, Michalski PJ, Wróblewski P, Tang Z, Kadlof M, Mazzocco G, Ruan Y, Plewczynski D
Note: '3D-GNOME: an integrated web service for structural modeling of the 3D genome' by SzalajP, Michalski PJ, Wróblewski P, Tang Z, Kadlof M, Mazzocco G, Ruan Y, Plewczynski D. NucleicAcids Res. 2016 May 16. pii: gkw437. pmid:27185892
2015
Authors: Tang Z, Luo OJ, Li X, Zheng M, Zhu JJ, Szalaj P, Trzaskoma P, Magalska A, Wlodarczyk J, Ruszczycki B, Michalski P, Piecuch E, Wang P, Wang D, Tian SZ, Penrad-Mobayed M, Sachs LM, Ruan X, Wei CL, Liu ET, Wilczynski GM, Plewczynski D, Li G, Ruan Y
Note: 'CTCF-Mediated Human 3D Genome Architecture Reveals Chromatin Topology for Transcription' Tang Z, Luo OJ, Li X, Zheng M, Zhu JJ, Szalaj P, Trzaskoma P, Magalska A,Wlodarczyk J, Ruszczycki B, Michalski P, Piecuch E, Wang P, Wang D, Tian SZ, Penrad-Mobayed M, Sachs LM, Ruan X, Wei CL, Liu ET, Wilczynski GM, Plewczynski D, Li G, Ruan Y.Cell 2015, Dec 17;163(7):1611-27. doi: 10.1016/j.cell.2015.11.024. Epub 2015 Dec 10.
2013
Abstract: 3D-Hit is a well established method for rapid detection of structural similarities between proteins, which is widely used in various bioinformatics web servers (MetaServer, GRDB, 3D-Fun, Rosetta, etc.). The algorithm decomposes proteins into set of overlaping segments of 9–13 residues, then tries to match them using root mean square distance metric. The best aligned pairs of segments are selected as seeds for futher analysis. Those initial hits are expanded by iterative process in order to construct the global structural alignment by concatenating pairs of matching segments. The method has the same accuracy as the other state-of-the-art structural comparison algorithms (LGscore2, DALI), yet it provides much faster processing times, and can be used in a high-throughput setup as the structural module of bioinformatics pipelines. The method is optimized in terms of speed and accuracy to work on novel computer architectures, such as PowerXCell8i and Sun Constellation System. Here, we provide the source code of the 3D-Hit program, describe selected architectures on which the software was ported, present programing models, point out significant porting steps and sumarize performance comparisons.
Authors: Ł Bieniasz-Krzywiec, Maciej Cytowski, L Rychlewski and D Plewczynski
Note: '3D-Hit: fast structural comparison of proteins on multicore architectures' by Ł. Bieniasz-Krzywiec, Maciej Cytowski, L. Rychlewski and D. Plewczynski. Optimization Letters (2013).